Мы извлекли их из базы данных, обозначив в графе Taxonomy "deltavirus" а в графе Description - "delta".
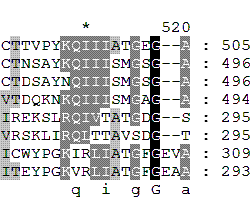
Глядя на последовательности, предположим два места возможного положения гэпов (буквально, что первым попалось на глаза).
Иллюстрация
{kind=link}
Множественное выравнивание, сделанное программой muscle *
Похоже, мы в какой-то степени угадали только со вторым гэпом, и всё равно программа сгруппировала остатки иначе.
Иллюстрация
{kind=link}
2.Множественное выравнивание белка AHPF_Ecoli и семи его вероятных гомологов (найдены с помощью программы Blast)
Q67QU3.1|FENR_SYMTH 49% сходства E-value 2e-22
Q2JFM8.1|FENR_FRASC 46% сходства E-value 3e-15
Q5HRY2.1|AHPF_STAEQ 70% сходства E-value 6e-148
P26829.1|DHNA_BACYN 75% сходства E-value 3e-173
P42974.2|DHNA_BACSU 56% сходства E-value 1e-166
P80880.3|TRXB_BACSU 55% сходства E-value 2e-45
P23160.1|R34K_CLOPA 54% сходства E-value 1e-29 выполненное программой muscle. *
Консервативные участки (самые яркие примеры)
- номера позиций выравнивания: 218-220; 222-225; 249-252; 326-328; 369-377; 392-394; 498-503
- номера соотв. позиций AHPF_Ecoli: 217-219; 221-224; 246-249; 318-320; 358-366; 381-383; 483-488
Самые близко расположенные небольшие консервативные участки можно объединять в один консервативный "блок".
Участков выравнивания, очевидно недостоверных с биологической точки зрения (с частыми гэпами и одиночными выравненными колонками), мало: как правило, если не выравнены все последовательности, найдены соответствия хотя бы в некоторых из них. Мне кажется подозрительным участок выравнивания в районе 302-312.

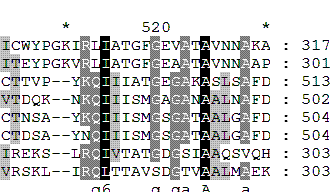
Гэпы неровные, похожие для некоторых белков участки проигнорированы. Этот фрагмент выравнивания неверен с биологической т.з. Выравнивание тех же последовательностей, сделанное программой mafft. *
В отличие от muscle здесь выравнены начальные метионины, что не имеет особого биологического смысла, а вот лишний гэп мы получаем. Спорный момент: mafft предпочла сопоставить друг другу глицины (обозначены чёрным), а не V, I и L, как сделала muscle. Хотя, не представляя структуры белка, мы можем только строить предположения. В данном конкретном случае глицин может быть важен, как "шарнир", и если мы построим выравнивание большего числа последовательностей и отметим эту же тенденцию, то это будет куда важнее с биологической точки зрения.
{kind=link}
{kind=link}
В целом консервативные участки те же.
Выравнивание в исполнении программы edialign пакета EMBOSS. *
Похоже на mafft за исключением кое-каких гэпов на участках с низкой консервативностью. Больше можно сказать об оформлении. Edialign пишет названия белков в самой таблице и выделяет разным регистром остатки в последовательности: заглавные буквы - собственно выравненные, а прописные - случайно попавшие в колонку при сдвигах и не претендующие на гомологичность.
3.Другие программы множественных выравниваний:
-consambig выделяет только консервативные остатки (разным регистром - большими буквами те, что встречаются во всех последовательностях, маленькими - в большинстве) - удобно, если ищешь именно консервативные позиции.
>EMBOSS_001 mxxxxxxkxqlxxxxzxxxxxxxxxxxxxxxxxsxxxxxlxxexaxxsxxxxxxxxxxxp vrxpsfxxxxxgxxxgxxfagxplghefxslvlajlxxxgxxxkxxqxxjxqxxxjxgxx xfexxxslxcxxcpdvvqalnxmsvjnpxixhtxidgxxxxxexxxxbxmxvpxvxxbgx xfxxgrmtxxxixxxxxxxxxkraaxexxxxxxxdXXXXGXGPXXXXXXXYXXXXXXXXX XXXXXxxxGGQXXXXypeXXXXBXXXXXXXXXXXXXXXXXXXXXXXXXXxXXXXxxxXxx xxxxxxXxXXXXXXXXXXXXXXXXXXXXXGXgxfXXXXXXXXXXXXxxXXXXXXXXXXXg XXXXXXXXXVXGGGBSXXXXXXXXXXXXXXXTXXXXXXXXXAXXXXXxXXXXXXXVXXXX XXXXXXXXXXXxXkXXXXXXXXXXXXXXXXXXXXXXXXXXGXXXXXgXXXXXXXXXxXXX XXXXBXXXXTXXXXXXAAGDXXXXXXXXXXXXXXXGXXAXXxxxxyxXXXxxxxxxxxxx xfpghssxxxxxg
-distmat выводит таблицу "эволюционных расстояний" для попарных выравниваний: в ячейках - количество замен на 100 остатков (ещё один способ оценки степени гомологии и построения эволюционных деревьев)
Distance Matrix --------------- Uncorrected for Multiple Substitutions Gap weighting is 0.000000 1 2 3 4 5 6 7 8 0.00 42.66 42.72 46.35 67.97 67.97 72.13 75.60 AHPF_ECOLI 1 0.00 16.31 35.97 69.58 66.45 72.90 75.84 DHNA_BACYN 2 0.00 36.76 67.44 67.76 72.33 76.04 DHNA_BACSU 3 0.00 67.77 66.67 75.00 75.35 AHPF_STAEQ 4 0.00 56.81 71.01 72.01 TRXB_BACSU 5 0.00 72.82 74.13 R34K_CLOPA 6 0.00 56.88 FENR_SYMTH 7 0.00 FENR_FRASC 8
-plotcon демонстрирует нам график среднего сходства по позициям (наглядно показывает самые "устойчивые" мотивы) *
{kind=link}