| Команда | Функция |
| fastqc chr10.fastq | Выдает информацию о качестве прочтений chr10.fastq |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr10.fastq trim_chr10.fastq TRAILING:20 MINLEN:50 | Выдает файл trim_chr10.fastq, где отрезаны с концов прочтений нуклеотиды качеством менее 20 и удалены чтения длиной меньше 50 |
| fastqc trim_chr10.fastq | Выдает информацию о качестве прочтений trim_chr10.fastq |
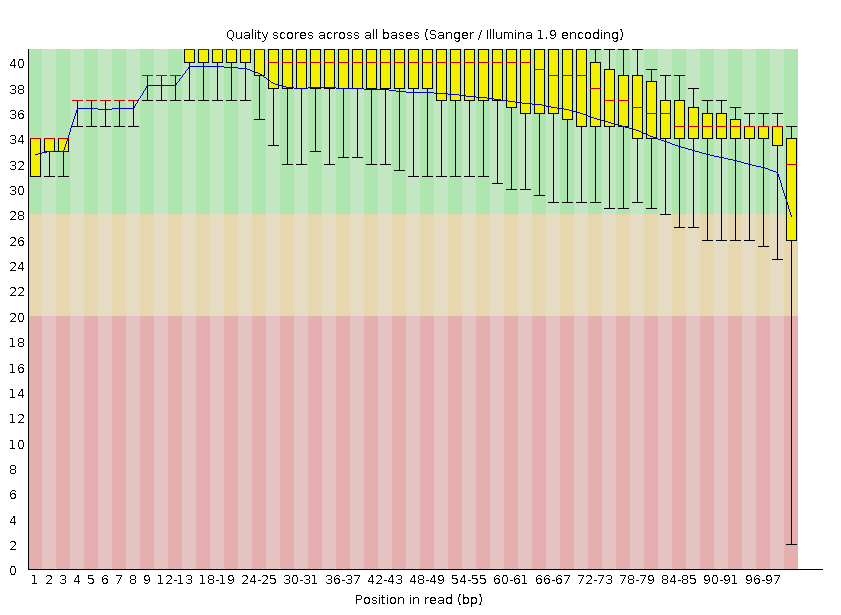 Рис. 1 Качество прочтения ридов до очистки
Рис. 1 Качество прочтения ридов до очистки
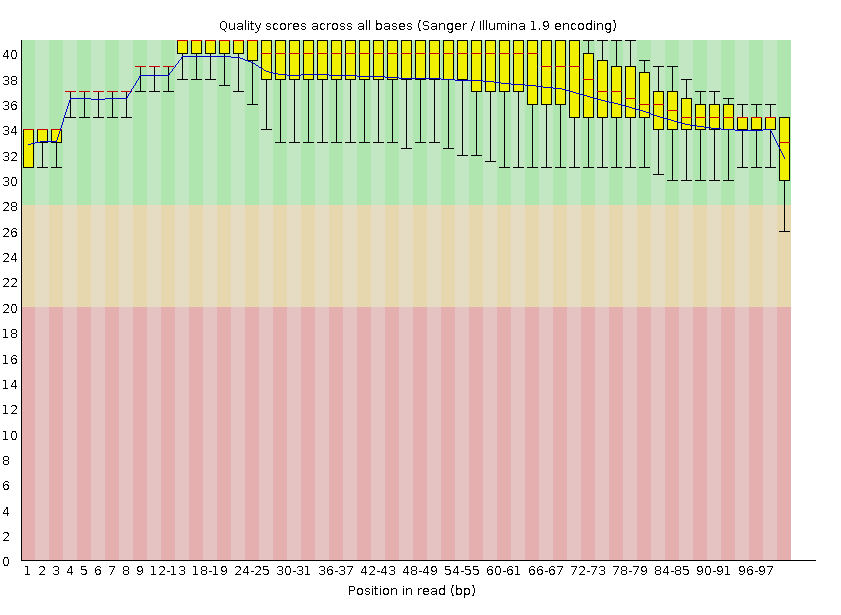 Рис. 2 Качество прочтения после очистки
До очистки файл содержал 10666 прочтений
После очистки файл содержал 10526 прочтений
Рис. 2 Качество прочтения после очистки
До очистки файл содержал 10666 прочтений
После очистки файл содержал 10526 прочтений
| Команда | Функция |
| hisat2-build chr10.fasta his_chr10 | Выдаёт индексированную референсную последовательность в формате fasta |
| hisat2 -x his_chr10 -U trimm_chr10.fastq --no-spliced-alignment --no-softclip > align.sam | Выравнивает прочтения с референсной последовательностью |
| samtools view align.sam -b -o align.bam | переводит файл из формата sam в формат bam |
| samtools sort align.bam -T file.txt -o alignsort.bam | Сортирует выравнивание чтений |
| samtools idxstats alignsort.bam > resut.txt | записывает число закартированных чтений |
| Команда | Функция |
| samtools mpileup -uf chr10.fasta sort.bam > snp.bcf | Создаёт файл с полиморфизмами |
| bcftools call -cv snp.bcf -o snp.vcf | Создаёт файл описания различий рефересной последовательности и данных прочтений |
| Координата | Тип мутации | Нуклеотид референса | Нуклеотид чтений | Глубина прочтения | Качество прочтения |
| 5781628 | Замена | T | G | 21 | 117.008 |
| 5781969 | Замена | A | T | 18 | 225.009 |
| 5784151 | Замена | A | G | 83 | 225.009 |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -o chr10.avinput |
| База данных | Команда |
| Refgene | perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr10_refg -build hg19 chr10.avinput /nfs/srv/databases/annovar/humandb/ |
| dbsnp | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -out chr10.snp -build hg19 chr10.avinput /nfs/srv/databases/annovar/humandb/ |
| 1000 genomes | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -out chr10_1000g -buildver hg19 chr10.avinput /nfs/srv/databases/annovar/humandb/ |
| GWAS | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -out chr10_gwas -buildver hg19 chr10.avinput /nfs/srv/databases/annovar/humandb/ |
| Clinvar | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -out chr10_clinvar -buildver hg19 chr10.avinput /nfs/srv/databases/annovar/humandb/ |