
Сравнение нуклеотидных последовательностей белков одного или же разных организмов является очень эффективным методом в области молекулярной биологии. В настоящее время, когда целые геномы секвенированы, поиски сходств последовательностей могут быть использованы для прогнозирования расположения и функции участков в ДНК, которые кодируют рассматриваемый белок регулируют его транскрипцию.
Basic Local Alignment Search Tool (BLAST) является наиболее часто используемым инструментом для расчета сходства последовательностей. BLAST позволяет использовать в сравнительном анализе различные базы данных. Кроме того, BLAST автоматически создает парное выравнивание анализируемой пользователем последовательности с найденной.
Возможности данного инструмента не ограничиваются представленными на этой странице. Все выполняемые программой и ее приложениями функции, а также информацию для их использования можно найти главной странице web-интерфейса к BLAST.
1. Поиск гипотетических гомологов изучаемого белка в разных банках
С помощью BLAST был проведен поиск гипотетических гомологов белка TENI_BACSU в трех банках данных:
• Swiss-Prot (SW)
• Protein Data Bank (PDB)
• Non-redundant protein sequences ("nr")
Таблица 1
Результаты поиска гипотетических гомологов белка TENI_BACSU
| Поиск по Swiss-Prot | Поиск по PDB | Поиск по "nr" | |
| 1. Лучшая находка (с последовательностью исходного белка) | |||
| Accession | P25053 | 1YAD_A | NP_389048.1 |
| E-value | 5e-146 | 2e-146 | 2e-144 |
| Вес (в битах) | 411 | 410 | 411 |
| Процент идентичности | 100% | 100% | 100% |
| 2. Число находок с E-value < 10–10 | 112 | 9 | 907 |
| 3. "Худшая из удовлетворительных" находка (последняя в выдаче с E-value < 1) | |||
| Номер находки в списке описаний | 378 | 24 | 4954 |
| Accession | Q8ZUF0.1 | 1KA9_F | ZP_07883970.1 |
| E-value | 0,98 | 0,65 | 1,00 |
| Вес (в битах) | 33,5 | 30,8 | 38,5 |
| % идентичности | 43 | 25 | 26 |
| % сходства | 56 | 45 | 41 |
| Длина выравнивания | 46 | 133 | 117 |
| Координаты выравнивания (от-до, в запросе и в находке) | В запросе:145-189 В находке: 243-288 |
В запросе:45-156 В находке: 75-204 |
В запросе:93-203 В находке: 96-212 |
| Число гэпов | 1 | 24 | 6 |
1. Лучшая находка (с последовательностью исходного белка)
На входе программы BLASTP подается код доступа (accession number) или последовательность в fasta-формате. По умолчанию поиск проводится по Swiss-Prot, выводится не более 100 найденных последовательностей и e-value <10. Эти данные могут быть изменены в параметрах алгоритма (Algorithm parameters).
Исследуемый белок был найден в Swiss-Prot и PDB. Так как “nr” включает в себя Swiss-Prot, то в нем белок был тоже найден.
2. Число находок с E-value < 10–10
Согласно таблице, наибольшее число явных гомологов наблюдается в “nr”. Это объясняется тем, что этот банк данных включает в себя все белковые последовательности из всевозможных источников.
Наименьшее число явных гомологов наблюдается в PDB, так как не для всех белков известны трехмерные структуры (именно их и содержит этот банк данных).
Аминокислотные последовательности известны для большего числа белков, поэтому число явных гомологов в Swiss-Prot гораздо больше, чем в PDB.
3. "Худшая из удовлетворительных" находка
(последняя в выдаче с E-value < 1)
Для поиска таких последовательностей был изменено количество выводимых найденных последовательностей (5000, вместо 100 по умолчанию).
Количество выводимых найденных последовательностей в “nr” при e-value<1 было ограничено (по умолчанию: 100). Предельный размер выдачи был увеличен до 20000. Однако, было найдено 4954 записи, так как было ограничение по e-value.
Поиск в Swiss-Prot и PDB ограничен параметрами по умолчанию: e-value<10 и предельный размер выдачи равен 100.
2. Поиск гипотетических гомологов изучаемого белка с фильтром по таксонам
С помощью BLAST можно найти гомологов белка TENI_BACSU в организмах других таксонов. Исследование можно провести по следующим таксонам:
- 'Eukaryota' (другое царство);
- 'Actinobacteria' (другой отдел того же царства бактерий);
- 'Clostridia' (другой класс того же отдела Firmicutes);
- 'Lactobacillales' (другой порядок того же класса Bacilli);
- 'Listeriaceae' (другое семейство того же порядка Bacillales);
- 'Geobacillus' (другой род того же семейства Bacillaceae);
- 'Bacillus anthracis' (другой вид того же рода).
Поиск ближайших гомологов проводится с ограничением по E-value<0,001. Чтобы найти наилучшего гомолога в организмах таксона, филогенетически как можно более далеко, проверку производим в порядке приближения к 'Bacillus subtilis'.
Таблица 2
Результаты поиска гипотетических гомологов белка TENI_BACSU в организмах других таксонов
| Поиск по Swiss-Prot | Поиск по PDB | Поиск по "nr" | |
| Eukaryota | |||
| Accession | Q5M731.1 | 3O05_A | CBK22495.2 |
| E-value | 8e-13 | 6e-04 | 2e-12 |
| Вес (в битах) | 68,9 | 38,9 | 72,8 |
| % идентичности | 29 | 43 | 30 |
| % сходства | 47 | 60 | 51 |
| Длина выравнивания | 211 | 56 | 213 |
| Координаты выравнивания (от-до, в запросе и в находке) | В запросе: 313-519 В находке: 3-200 |
В запросе: 193-248 В находке: 137-189 |
В запросе: 240-243 В находке: 3-205 |
| Число гэпов | 17 | 3 | 19 |
В трех банках данных гомологи были обнаружены в 'Eukaryota' (другое царство).
3. BLAST двух последовательностей
При изменении параметров на входе программы можно подать две последовательности (например, TENI_BACSU и наиболее идентичный ему 3O05_A из другого организма). В этом случае алгоритм сделает парное выравнивание заданных последовательностей. Кроме того, будет получена и карта локального сходства заданных последовательностей (для этого нужно открыть Dot Matrix View):
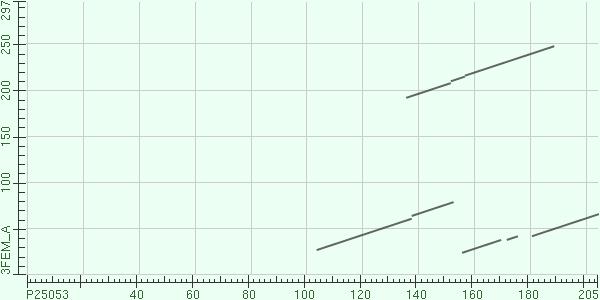
Рис.1. Карта локального сходства при
E-value = 10 (по умолчанию)
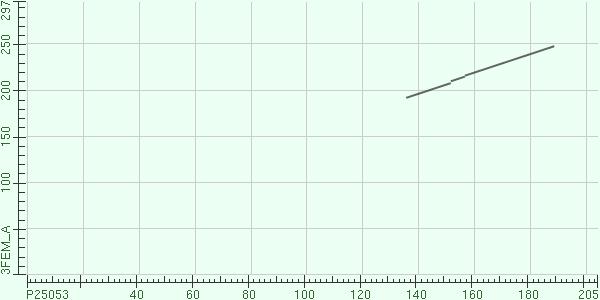
Рис. 2. Карта локального сходства при E-value = 0,01
E-value, как видно из рисунков, влияет на карту локального сходства. Этот параметр пересчитывается из веса локального выравнивания и длин сравниваемых последовательностей. BLAST находит локальные выравнивания, у которых E-value ниже заданного порога, и отображает их на карте.
При E-value = 10 на Рис.1 семь локальных выравниваний. Если уменьшить порог E-value, то некоторые из найденных выравниваний при более высоком пороге теряются, что видно на Рис. 2 (только два локальных выравнивания). Это значит, что остальные пять не представленные на Рис. 2 выравнивания имели 0,01< E-value <10.