Парное выравнивание белков – это ключевой метод биоинформатики, заключающийся в сопоставлении двух белковых последовательностей одинаковой длины, допускающего вставку в обе последовательности специального знака пропуска (гэпа, от англ. gap), обозначающегося символом «-» или «•».
Так как последовательности белков и нуклеиновых кислот являются следствием эволюции, то совпадение на большой протяженности, вероятно, отражает эволюционное родство между белками, а иногда сходство их активностей и функций. Однако, возможно и совпадение мотивов (коротких участков белка, типичных для какой-либо определенной активности) между очень разными белками, которые выполняют различные функции.
Очевидно, что выравнивание (как и любое сопоставление) может быть «удачным» и «неудачным».
«Удачное» выравнивание не является случайным. Это возможно благодаря механизму репликации, в котором участвует ДНК-полимераза, обеспечивающая точность при копировании. Однако, этот фермент тоже может совершать ошибки, которые исправляются системой репарации. Но даже это не обеспечивает 100% точности копирования.
Программам, которые строят выравнивания, требуется численная мера для оценки качества сопоставления. Измерить «удачность» выравнивания можно:
- По % совпадения (англ. identity )
- используется для оценки качества уже построенного выравнивания
- По % сходства (англ. similarity )
- аминокислоты группируются по природе бокового радикала
- Используя специфические весы на замену каждой пары аминокислотных остатков
1. Модель эволюции белковых последовательностей
Скрипт evolve_protein.pl позволяет получить фрагменты из искусственно смоделированных мутантов белков.
У скрипта два обязательных параметра:
- -i: указывает с последовательностью изучаемого белка в fasta-формате;
- -o: указывает имя файла с результатом работы скрипта.
Скрипт моделирует эволюцию последовательности без учета естественного отбора, то есть отображает ошибки в работе ДНК-полимеразы и системы репарации./p>
Для каждой буквы последовательности есть вероятность (change ), что в данной позиции случится ошибка. Параметр –с задает такую вероятность. по умолчанию скрипт работает с change =0.2. Это означает, что в среднем в 20% букв будет происходить изменение, а в 80% - нет.
Если изменение в позиции произошло, то есть вероятность, что аминокислота этой позиции была заменена. Параметр –r задает вероятность замены (replace ). По умолчанию скрипт работает с replace =0.5. Если замена не произошла, то с вероятностью 0,5 случится делеция (удаление аминокислоты из последовательности), и с вероятностью 0,5 – инсерция (вставка случайной аминокислоты перед той, что рассматривается).
Существуют и другие опции скрипта:
- -f: создание полноразмерной последовательности;
- -t: создание полноразмерной последовательности;
- -g: создание полноразмерной последовательности;
С помощью скрипта evolve_protein.pl были 3 коротких фрагментов (по 20 аминокислот) из искусственно смоделированного мутанта белка TENI_BACSU:
-
>0|simulation_result|change=0.6|replace=0.6|generations=1
RDPAAFSTKRRHKLPNKISF
Полный результат работы скрипта -
>0|simulation_result|change=0.6|replace=0.8|generations=1
NSHLSKIFTRIEVMAGKATY
Полный результат работы скрипта -
>0|simulation_result|change=0.4|replace=0.8|generations=1
YAVGRIKECAYLAGKVATPC
Полный результат работы скрипта
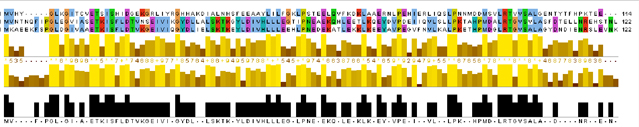
Наглядно выравнивания можно проиллюстрировать с помощью JalView – интегрированного с JMol многофункционального браузера выравниваний.
JalView позволяет группировать аминокислоты по природе бокового радикала с помощью цветовой гаммы. В ниже представленных выравниваниях аминокислоты разделены на группы по цвету следующим образом:
- с отрицательно заряженным радикалом – красный;
- с положительно заряженным радикалом – синий;
- с ароматическим радикалом – фиолетовый;
- с радикалом, в состав которого входит сера – серый;
- с незаряженным полярным радикалом – светло-зеленый;
- с алифатическим радикалом – желтый;
- иминокислота пролин (Pro) – розовый.
С его помощью получены парные выравнивания последовательности белка TENI_BACSU и участков последовательностей мутантов, для которых были посчитаны % совпадения (identity ), % сходства (similarity ) вес по матрице BLOSUM62 (alignment weight ).
-
Мутант 1 (change=0.6|replace=0.6)

Identity: 9/21*100% = 42,8% Similarity: 12/21*100%= 57,1% Alignment weight: 26
-
Мутант 2 (change=0.6|replace=0.8)
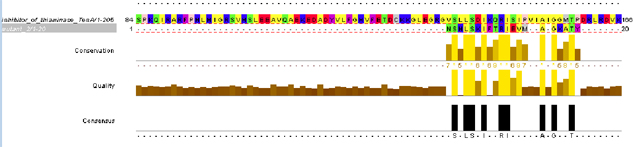
Identity: 9/23*100% = 39,1% Similarity: 11/23*100%= 47,9% Alignment weight: 16
-
Мутант 3 (change=0.4|replace=0.8)

Identity: 11/23*100% = 47,9% Similarity: 12/23*100%= 52,2% Alignment weight: 28
Выравнивания 1 и 2 более или менее сходны по % совпадения и % сходства, так для них change =0.6. При уменьшении этого параметра большее количество аминокислот остались не измененными в своей позиции, что привело к увеличению % совпадения и % сходства.
На примере выравниваний 1 и 2 можно увидеть, как влияет параметр replace : при увеличении значения replace , процент сходства уменьшается.
Различия процентов сходства, вероятно, является не самым объективным параметром для сравнения, так как разделение аминокислот по свойствам бокового радикала достаточно условно и сильно зависит от условий конкретной задачи.
2. Выравнивание TENI_BACSU его ортологов
JalView был использован для построения и анализа выравнивания последовательности белка TENI_BACSU и двух его ортологов Q73DB4_BACC1 и A7Z3F5_BACA2.
С помощью программы Muscle были выровнены все три последовательности. Для каждого из трех парных выравниваний была получена информация с помощью команды infoalign пакета EMBOSS.
Таблица 1
Характеристика выравнивания последовательностей белка TENI_BACSU и Q73DB4_BACC1
| Name | Sequence length | Aligned length | Identify | Similarity | Difference | Gaps | % change | Weight |
| TENI_BACSU | 205 | 205 | 205 | 0 | 0 | 0 | 0.000000 | 1.000000 |
| Q73DB4_BACC1 | 206 | 207 | 90 | 45 | 71 | 1 | 56.521740 | 1.000000 |
Таблица 2
Характеристика выравнивания последовательностей белка TENI_BACSU и A7Z3F5_BACA2
| Name | Sequence length | Aligned length | Identify | Similarity | Difference | Gaps | % change | Weight |
| TENI_BACSU | 205 | 205 | 205 | 0 | 0 | 0 | 0.000000 | 1.000000 |
| A7Z3F5_BACA2 | 205 | 205 | 141 | 31 | 33 | 0 | 31.219513 | 1.000000 |
Выравнивание всех трех последовательностей было окрашено по схеме CrustalX. Опция Above identity ntreshold позволяет окрашивать только те позиции, в которых идентичны как минимум две аминокислоты (порог идентичности 67%)