
последовательностям. Деревья, содержащие паралоги. Отредактировано 22/09/13
Реконструкция филогенетических деревьев по нуклеотидным последовательностям. Деревья с паралогами
Для построения филогенетического дерева по последовательностям 16S rRNA (РНК малой субъединицы рибосомы) выбранных ранее бактерий (см. Практикум 2, Практикум 3) были найдены последовательности 16S rRNA каждой из бактерий (Таблица 1). Для этого из записей EMBL с полными геномами программой –seqret были вырезаны необходимые участки.
Участки РНК малых рибосомных субъединиц выбранных бактерий
| Название бактерии | Мнемоника/td> | AC записи EMBL | Начало | Конец | Направление |
| Bacillus subtilis | BACSU | AL009126 | 30279 | 31832 | + |
| Enterococcus faecalis | ENTFA | AE016830 | 248466 | 249987 | + |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421 | 11973 | + |
| Clostridium tetani | CLOTE | AE015927 | 176113 | 177621 | + |
| Listeria monocytogenes | LISMO | AL591974 | 37466 | 39020 | + |
| Lactobacillus delbruecki | LACDA | CR954253 | 45160 | 46720 | + |
| Staphylococcus epidermidis | STAES | AE015929 | 1598006 | 1599559 | - |
Полученные последовательности были выровнены с помощью программы JalView (выравнивание).
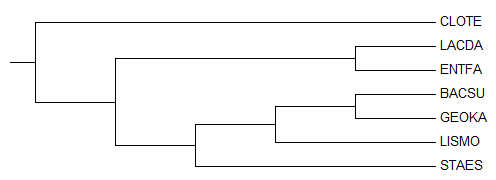
Рис. 1 Правильное филогенетическое дерево
В программе Mega методом Neighbor-Joining Using BLOSUM62 было построено дерево (Рис. 2) на основе полученного выравнивания.
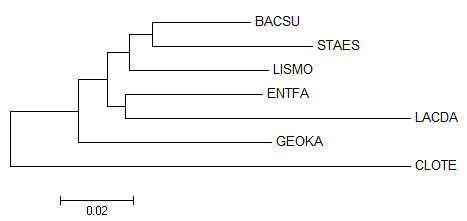
Рис. 2 Филогенетическое дерево, построенное по нуклеотидным последовательностям.
Сравнивая полученное дерево (Рис.2) с правильным (Рис.1), можно отметить, что только весть {LACDA, ENTFA} vs {CLOTE, BACSU, GEOKA, LISMO, STAES} является для них общей.
Есть несколько способов повлиять на результат.
Во-первых, использовать бутстреп-анализ (Рис.3).
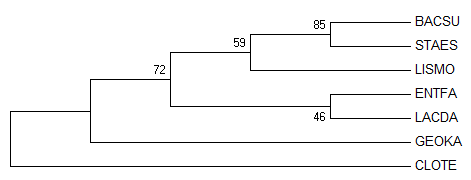
Рис. 3 Филогенетическое дерево, построенное по нуклеотидным последовательностям методом Neighbor-Joining с использованием бутстреп-анализа
Такой способ не приблизил полученное дерево к правильному (например, по количеству общих нетривиальных ветвей).
Во-вторых, можно удалить из исходного для построения дерева выравнивания концевые участки (10 нуклеотидов в начала и 35 нуклеотидов в конце), которые оказывали значительное влияние на результат, потому что есть не во всех последовательностях. Для редактированного выравнивания было построено дерево (Рис. 4)
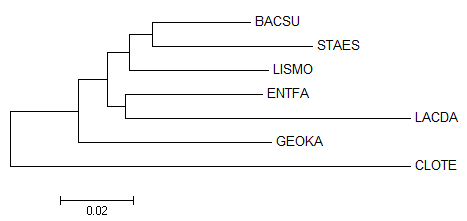
Рис. 4 Филогенетическое дерево, построенное по редактированному выравниванию методом Neighbor-Joining
Такой способ не приблизил полученное дерево к правильному (например, по количеству общих нетривиальных ветвей).
Следовательно, можно сказать, что качество построения деревьев по нуклеотидным последовательностям в данном случае ниже, чем построение по белковым последовательностям.
Для построения дерева достоверных гомологов белка CLPX_BACSU в выбранных бактериях был использован файл proteo.fasta, который содержит записи банка UNIPROT, относящиеся к рассматриваемым бактериям. Для нахождения гомологов была использована программа blastp, а затем по мнемонике выбранных бактерий был отобраны соответствующие им белки. Выравнивание полученных гомологов было выполнено программой MUSCLE.
С помощью программы Mega методом Neighbor-Joining Using BLOSUM62 было получено дерево, реконструированное по выравниванию гомологов белка CLPX_BACSU в выбранных бактериях (Рис.5).

Рис. 5 Филогенетическое дерево, построенное по выравниванию гомологов белка CLPX_BACSU методом Neighbor-Joining
Будем считать, что два гомологичных белка являются ортологами, если они из разных организмов, и разделение их общего предка на линии, ведущей к ним, произошло в результате видообразования. Два гомологичных белка из одного организма будем называть паралогами.
Тогда на полученном дереве (Рис.5) есть ортологи (например, HSLU_LACDA и HSLU_ENTFA, CLPX_LISMO и CLPX_BACSU) и паралоги (например, Q1G869_LACDA и Q1GBM8_LACDA, Q891B9_CLOTE и Q899H3_CLOTE).