Для сравнения наборов систем рестрикции-модификации была выбрана бактерия Oscillibacter valericigenes (AP012044.1). Вот ссылки на полный геном и набор контигов из метагенома.
Oscillibacter_valericigenes.fasta.gz
Использовался данный в задании список сайтов, узнаваемых ферменты систем рестрикции-модификации.
С помощью веб-сервиса (опция “алгоритм Карлина”) для генома из NCBI было получено 16 сайтов, для которых контраст меньше 0,78. Для метагенома было найдено 18 сайтов (wgs файлы). Файл с результатами: chr.tsv и wgs.tsv Файл с отобранными сайтами: chr_sorted.tsv и wgs_sorted.tsv Если посмотреть на цифры “observed_number”, то видно, что в хромосоме в целом больше сайтов. Далее был произведен сравнительный анализ недопредставленных сайтов - в таблице были отмечены цветом сайты, встречающиеся только в метагеноме, только вхромосоме и пересекающиеся all_sorted.xls. Можно сказать, что многие сайты встречаются только в метагеноме.Некоторые встречаются только в хромосоме. Пересекается примерно половина недопредставленных сайтов.
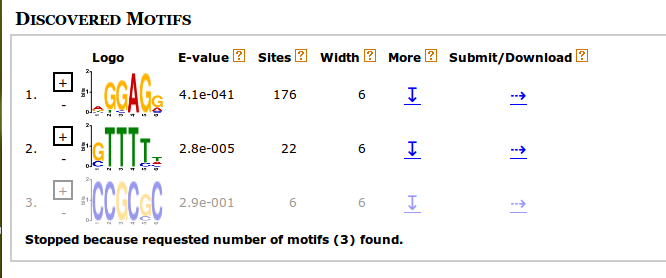
Сначала с сервера NCBI была скачана кольцевая хромосома. Затем были выбраны 300 генов - не hypothetical protein и не слишком короткие. Для каждого гена с помощью скрипта был вырезан участок от -16 до 0 позиции до начала кодирующей последовательности. С помощью MEME были найдены мотивы в вырезанных фрагментах. Искались мотивы длиной от 4 до 6, 0 или 1 мотив на последовательность, только на одной цепи. Сначала были найдены три мотива, чтобы сравнить результаты и e_value. Рис 1 Результат работы МЕМЕ
Первый мотив значительно более достоверный чем остальные, поэтому он скорее всего является искомой послдовательностью SD. Затем был произведен поиск одного мотива, и была найдена позиционно-весовая матрица PWM.txt. На основе позиционно-весовой матрицы был произведен поиск найденного мотива для всех остальных генов бактерии. Для всех генов были вырезаны участки от -16 до 0 позиции. Было найдено 2849 мотивов для p-value меньше 0.01. Возможно, присутствует большое количество случайных находок. Вот файл FIMO_table_0.01.xls, в котором результат работы FIMO и гистограмма. Рис 2 Гистограмма распределения начала найденных SD от страта трансляции По оси Х позиция от начала трансляции, по оси У количество генов.
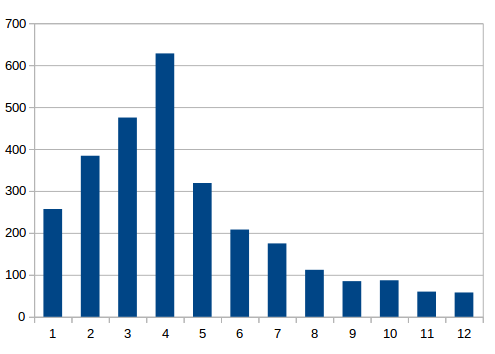
Видно, что большее количество SD начинаются с 4 нуклеотида от начала трансляции. Также для мотивов был построен LOGO с поомощью одноименного сервиса. Рис 3 Лого, построенное по мотивам с порогом 0.01
Качество данных было проверено с помощью FastQC. Вот результат: task_3/chipseq_chunk49_fastqc.html На графике можно видеть, что качество плохое, поэтому файл был обработан с помощью trimmotanic командой : java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk49.fastq outfile_35.fastq SLIDINGWINDOW:10:35
Затем чтения были откартированы на геном человека hg19 с помощью команды
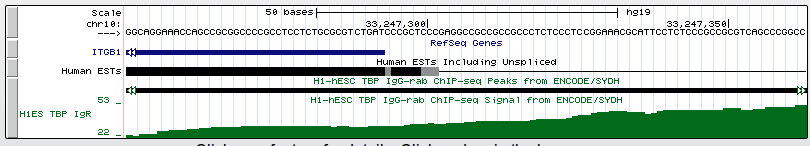
bwa mem srv/databases/hg19/GRCh37.p13.genome.fa outfile_35.fastq > outfile_35.sam. samtools view -bSo outfile_35.bam outfile_35.sam samtools sort outfile_35.bam -T chip_temp -o outfile_35.sorted.bam samtools index outfile_35.sorted.bam samtools idxstats outfile_35.sorted.bam > outfile_35.idxstats samtools view -c outfile_35.sorted.bam Всего было 17402 чтений, все они были откартированы на геном. При этом распределение чтений по хромосомам позволяет предположить, что это хромосома 10. task_3/output_35.idxstatsДалее был произведен поиск пиков помощью программы MACS. Использовалась команда macs2 callpeak -t outfile_35_sorted.bam -n chipseq_chunk49 --nomodel. Были получены следующие файлы: outfile_35_peaks.narrowPeak, outfile_35_peaks.xls и outfile_35_summits.bed. Всего найдено 9 пиков. Ни один из них не лежит в области гена. Их расположение можно посмотреть на рисунках:
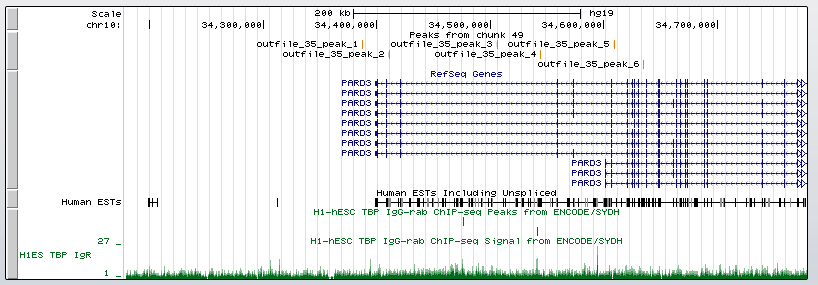
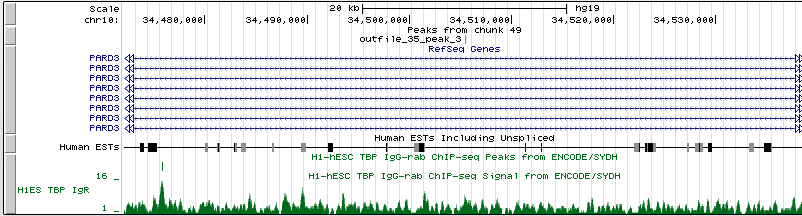
Здесь можно посмотреть координаты каждого из пиков:
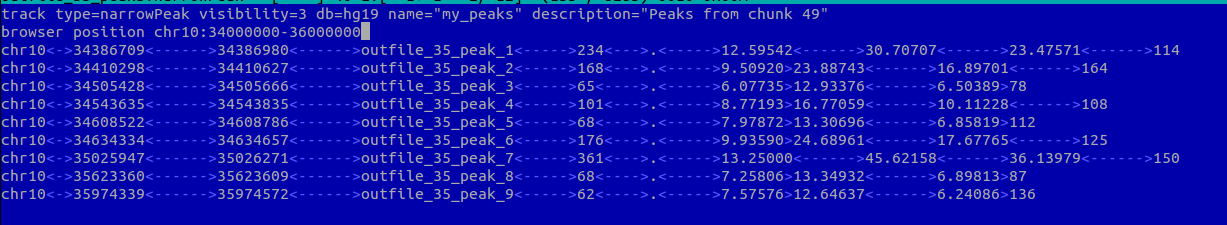
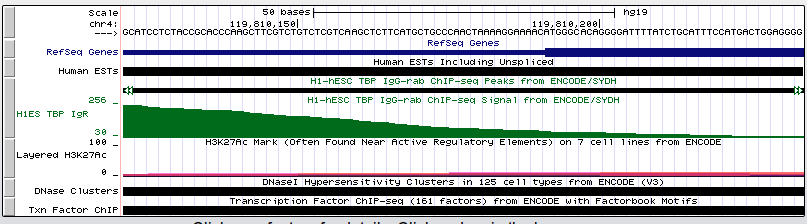
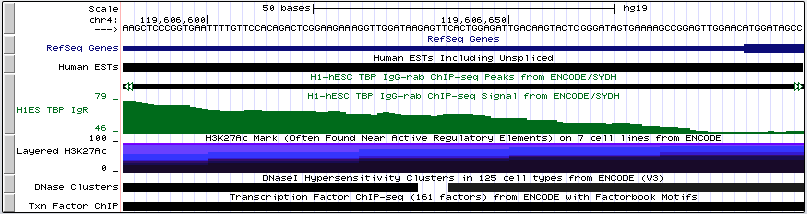
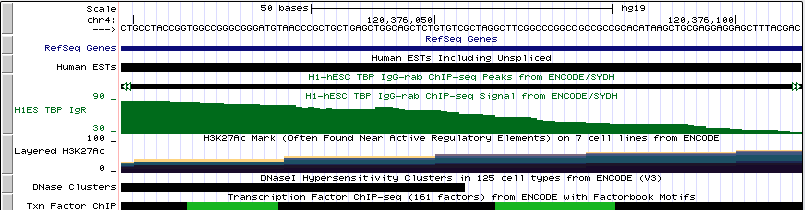
С помощью геномного браузера UCSC были проанализированы результаты ChIP-seq анализа для TBP в клеточной линии H1-hESC. Были найдены три гена, перед которыми есть ТАТА бокс, и один перед которым ТАТА бокса нет. Далее приведены сведения о наличии-отсутствии ТАТА бокса и изображения расположения генов.