Анализ транскриптомов.
1. Анализ качества чтений.
При помощи программы FastQC был проведен контроль качества ридов.
Её результат был выдан в качестве страницы html.
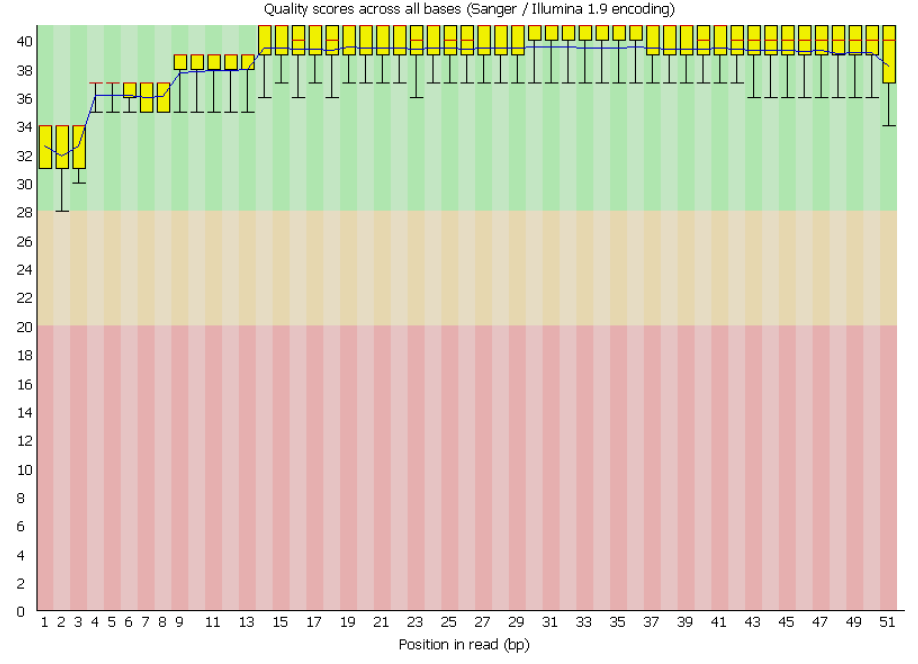
Как видно из изображения выше, качество прочтений отличное. Q > 30 на всем участке и даже хвосты не выходят за пределы зеленой зоны,
которая отграничивает хорошие прочтения от допустимых. Передний конец имеет немного хуже качество, однако и оно считается хорошим.
2. Картирование чтений.
Файл выданной программой Hisat2
Вызванные команды:
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
hisat2-build chr11.fasta chr11 (Этот шаг был проделан в пр.11 и использовались те же индексные файлы)
hisat2 -x chr11 -U chr11.1.fastq -S pr12_his.sam --no-softclip
Убрали параметр --no-spliced-alignment, так как при картировании транскриптомов нужно разрешить разрывы.
Это связано с тем, что мог происходить сплайсинг.
3. Анализ выравнивания.
| Вызванные команды: | Что делают: |
| samtools view -b pr12_his.sam -o pr12_ex4.bam | Переведение выравненных с референсом чтений(файл ex3.sam) в бинарный формат .bam |
| samtools sort pr12_ex4.bam -T pr12.txt -o pr12_sort.bam | Сортировка выравненных с референсом чтений(в .bam формате) по координате в референсе начала чтения |
| samtools index pr12_sort.bam | Индексация отсортированного .bam файл |
| samtools idxstats pr12_sort.bam > out_pr12.txt | Показывает сколько чтений откартировано на геном |
Из 39549 откартировано на геном 39269. 280 прочтений не откартировались.
4. Подсчет чтений.
| Вызванные команды: | Что делают: |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtobed -i pr12_sort.bam > reads.bed | Перевод файла *.bam в *.bed, туда кладутся координаты каждого выровненного чтения |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b reads.bed -c > overlap.bed | Пересечение разметки генов с полученными координатами, результат выдается в файл *.bed |
| sort -k 6 -r overlap.bed > sort_overlap.bed | Для простоты анализирования полученных результатов, файл был отсортирован |
5. Анализ результатов
Как видно из отсортированного файла sort_overlap.bed, в основном все прочтения легли в границы гена HSPA8.
Этот ген кодирует белок из семейства белков теплового шока 70(Hsp70). Его ориентировочная масса составляет 70 кДа. Он функционирует как шаперон связывается с зарождающимися полипептидами для облегчения правильного фолдинга. Он также функционирует как АТФаза при разборке везикул, покрытых клатрином, при транспортировке мембранных компонентов через клетку.
Также небольшие фрагменты попали на гены малых ядрышковых РНК.
6. Дополнительные задачи по Bedtools.
| Вызванные команды: | Что делают: |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtofastq -i pr12_sort.bam -fq reads_2.fq | Получает из файла с выравниванием(pr12_sort.bam) файл с чтениями(reads_2.fq) в формате fastq |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools getfasta -fi chr11.fasta -bed reads.bed > bedtools_2.fasta | Получает файл с нуклеотидной последовательностью (chr11.fasta) для одного из покрытых чтениями генов |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools cluster -i reads.bed > clust.bed | Объединяет чтения в кластеры
(Было получено 30 кластеров) |