Для выполнения задания были выбраны бакрерии Brucella abortus 104M (chromosome 1, complete sequence) и Brucella suis bv. 2 strain Bs143CITA (chromosome I, complete sequence).
Карта локального сходства отражает крупные эволюционные события, такие как вставки, делеции, транслокации, инверсии. Она отбражает информацию о том, какой участок генома одной бактерии соответствует определенному участку генома другой бактерии.
Чтобы сравнить геномы двух бактерий, было нужно запустить алгоритм blastn для выравнивания 2 последовательностей (blast2seq) с сайта NCBI.
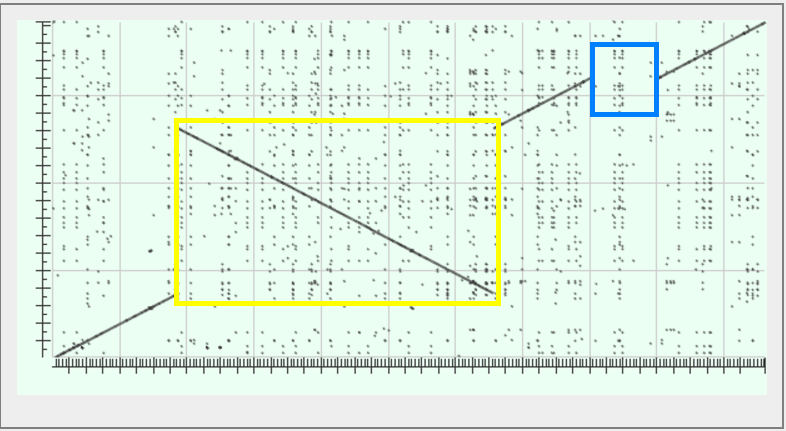
На карте локального сходства отображены следующие особенности:
- Жёлтой рамкой выделена инверсия участка.
- Голубой рамкой выделено место, в котором либо произошла вставка, либо делеция. Т. к. длина хромосомы Brucella abortus 104M длиннее, чем у Brucella suis bv. 2 strain Bs143CITA, то скорее всего могла произойти вставка в последовательность Brucella abortus 104M или делеция в хромосоме Brucella suis bv. 2 strain Bs143CITA.
Задание2. Описание сходств и различий геномов близкородственных бактерий.
Для выполнения задания использовался пакет NPG-explorer, установленный на kodomo, с помощью которого было сделано построение нуклеотидного пангенома (NPG). Нуклеотидный пангеном - это специальный формат для множественного выравнивания геномов, ориентированный на геномы близкородственных бактерий или архей.
Для выполнения этого задания я выбрала 3 штамма вида Helicobacter pylori: F16, F57 и F32.
Для работы пакета NPG-explorer было необходимо создать файл genomes.tsv, в котором содержится информация о том, откуда брать последовательности геномных ДНК и аннотации генов.
Затeм с помощью команды: npge -g npge.conf был создан файл npge.conf. , содержащим параметры программы.
Был изменён параметр MIN_IDENTITY на рекомендованное программой значение 0.877.
Информация о блоках в формате Excel
Выравнивание:





По выравниванию видно, что 6 блоков у всех штаммов расположены на одном и том же месте (10, 14, 22, 26, 36, 39).
Краткая справка по терминологии:
s-блоки - стабильные (коровые) блоки, по одному фрагменту из каждого генома
h-блоки - "полустабильные" блоки - по одному фрагменту из части геномов
u-блоки - и не блоки вовсе, а уникальные последовательности из одного генома, у них нет гомологов среди всех геномов, кроме самой себя
r- блоки - блоки с повторами, по крайней мере, в одном геноме
m-блоки - минорные блоки - короткие ( Список глобальных блоков - синтений - см. в global-blocks/blocks.gbi g-блоки (глобальные блоки) состоят из последовательно идущих во всех геномах s-блоков, перемежающихся блоками других типов (r-, h-, u- и m-).
Описание g-блоков (синтеничных участков).
Использовался файл blocks.blocks, из которого бралась информация по G-блокам.
Описание s-блоков (стабильных).
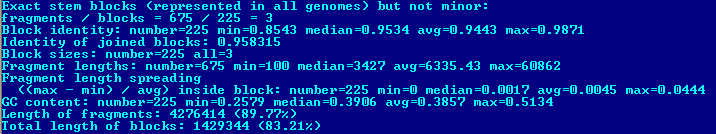
Данные из файла pangenome.info
Описание повторов на примерах r-блоков.
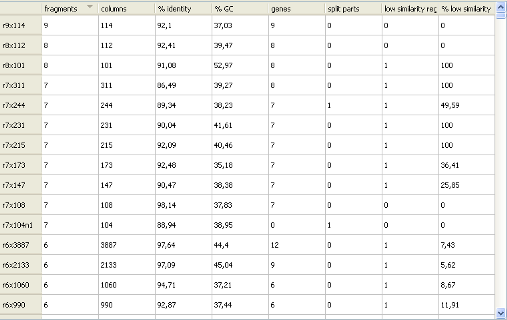
Блок r7x147 - - тип блока (от repeat), в блоке 7 фрагментов, 147 позиций в выравнивании блока.

выравнивание последовательностей, составляющих данный блок:
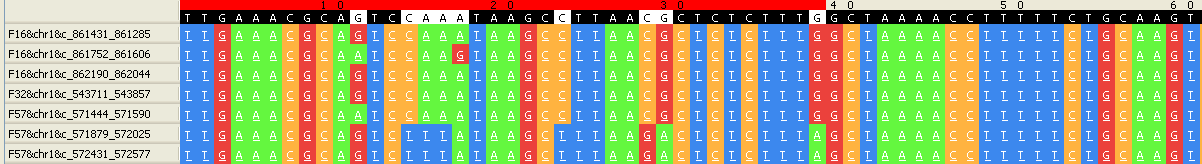
Этот блок содержит в совокупности фрагменты 7 генов.
Другой повтор - блок r6x134

выравнивание последовательностей, составляющих данный блок:
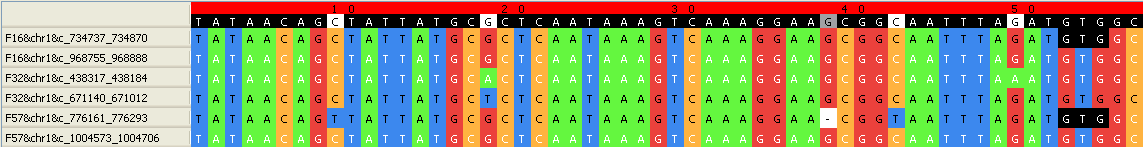
Этот блок содержит в совокупности фрагменты 5 генов.
h-блоки
H-блоки - это "полустабильные" блоки, включающие по одному фрагменту из части геномов. То есть у какого-то из организмов данная последовательность отсутствует.
Ниже представлены 4 самых длинных h-блока:
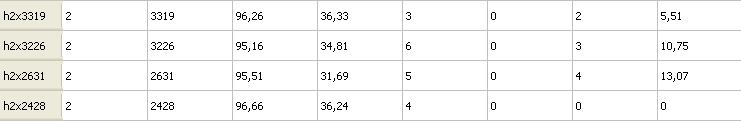
Они соответствуют четырём крупным делециям.
u-блоки
Примеры уникальных последовательностей:
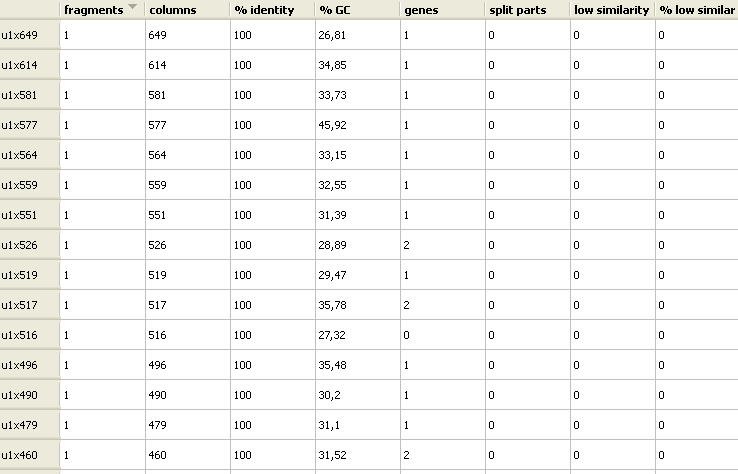
Блок u1x112, есть у штамма F16.
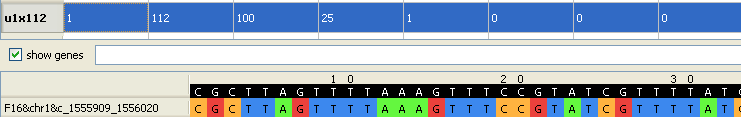
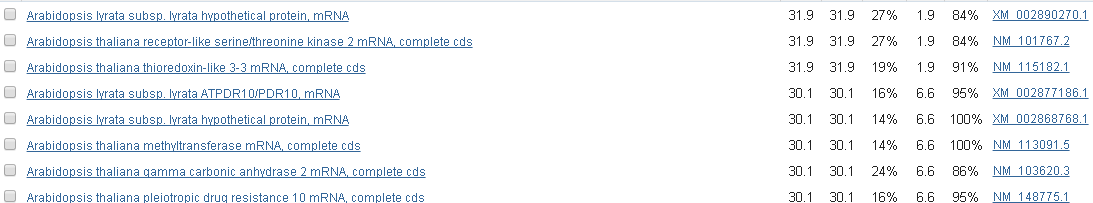
Белок содержит 1 ген. Блок u1x112 содержит последовательность, гомологичную гену С/Т киназы мРНК, , найденнй в других штаммах данного вида (рис.).
СПАСИБО ЗА ПРОСМОТР
© Мария Медведева