Часть I: подготовка чтений
Анализ качества чтений
Анализ и очистка чтений проводились для файла
chr13.fastq.
С помощью программы
FastQC проведён контроль качества скачанных чтений.
Отчёт программы можно увидеть здесь.
| В отчёте программы FastQC можно найти информацию о исследуемых чтениях. В скачанном файле используется кодировка Sanger / Illumina 1.9.
Всего файл содержит 12155 чтений. Последовательности имеют длину от 30 до 100 нуклеотидов. Чтения содержат 41% глутамина и цитозина. |
Очистка чтений
Была проведена очистка чтений с помощью программы Trimmomatic.
Адаптеры уже были удалены. С каждого конца прочтения отрезаны нуклеотиды с качеством ниже 20 (опция TRAILING:20),
удалены прочтения длиной меньше 50 нуклеотидов (опция MINLEN:50). При запуске программы указан phred33 формат fastq (для Sanger / Illumina 1.9).
В итоге, для запуска Trimmomatic использовалась команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq trimm.fastq TRAILING:20 MINLEN:50
В результате получен новый файл с чтениями - trimm.fastq, который снова был проанализирован с помощью FastQC. Отчёт по очищенным чтениям пожно увидеть
здесь.
|
Отчёт FastQC отличается от аналогичного для файла с неочищенными чтениями. Теперь файл содержит 11933 чтений
(количество уменьшилось не сильно, что говорит о том, что чтения изначально были довольно хорошими). Длина чтений теперь варьирует от 50 до 100
нуклеотидов, т. к. сначала чтения были обрезаны, а потом удалены те из них, которые имели длину меньше 50.
Процентное содержание гуанина и цитозина осталось неизменным. |
Оценка
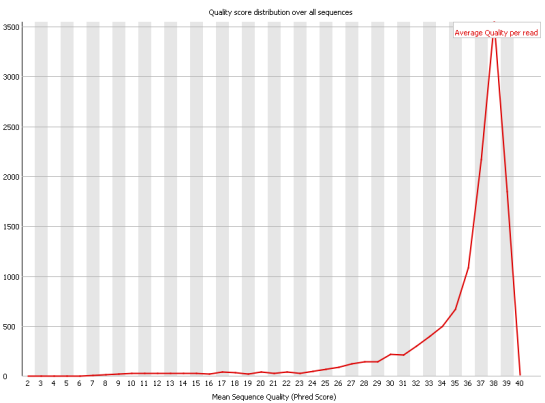
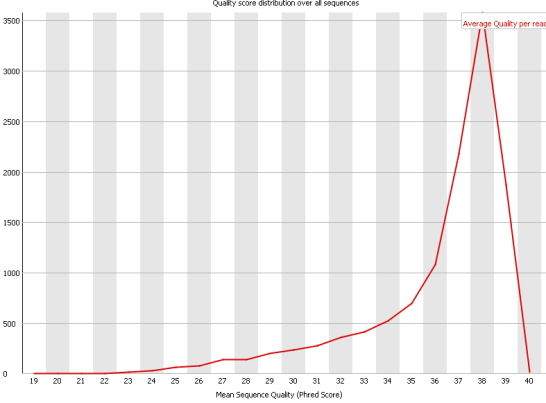
Качество нуклеотидов (т. е. точность определения нуклеотида в каждой позиции) в целом возросло, но не очень значительно.
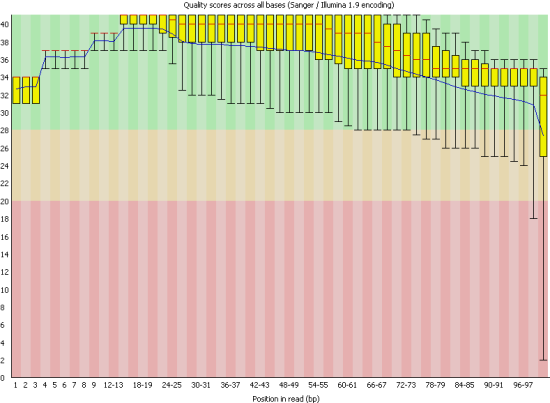
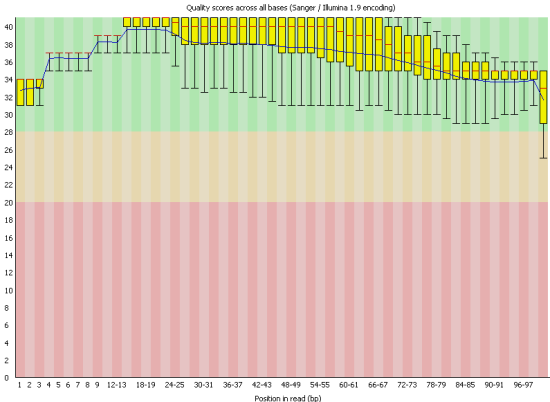
Это отражает график Per base sequence quality. Здесь представлено сравнение исходного и улучшенного с помощью программы Trimmomalic графики.
До обработки Trimmomatic на графике были нуктеотиды, минимальная оценка качества которых попадала в красную зону, после улучшения их качество заметно
улучшилось.
Графики, отражающие качество каждого нуклеотида в чтениях (Per base sequence quality). Слева приведён график для исходного файла с чтениями,
справа - график после улучшения с помощью программы Timmomatic. Графики получены с помощью программы FastQC.
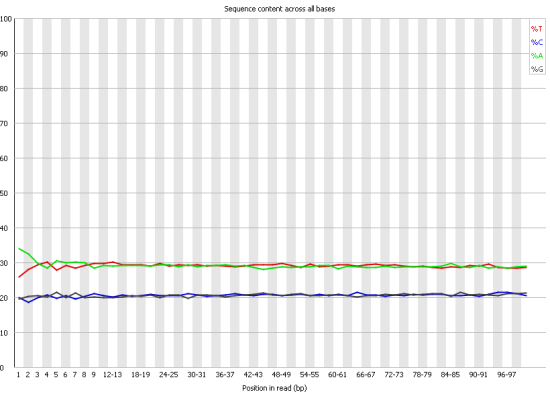
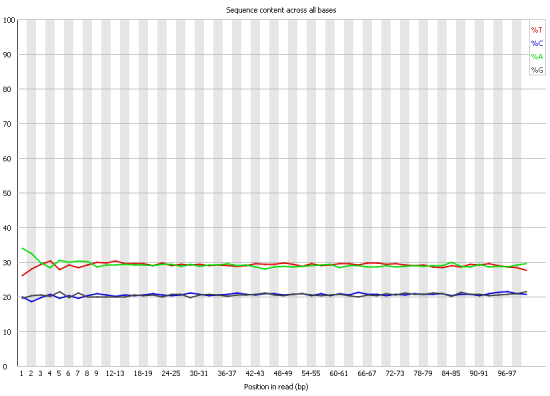
График, отражающий частоту встречаемости каждого из четырёх нуклеотидов (Per base sequence content) совсем не изменился.
Графики, отражающие распределение длин чтений (Per base sequence content). Слева приведён график для исходного файла с чтениями,
справа - график после улучшения с помощью программы Timmomatic. Графики получены с помощью программы FastQC.
Совсем немного изменилось распределение длин чтений. На графиках Sequence length distributionможно увидеть, что до улучшения все чтения
имели длину от 30 до 100, а после улучшения - от 50 до 100. Однако, всё равно почти все чтения имеют длину 100 нуктеотидов, количество более коротких чтений незначительно.
Графики, отражающие распределение длин чтений (Sequence length distribution). Слева приведён график для исходного файла с чтениями,
справа - график после улучшения с помощью программы Timmomatic. Графики получены с помощью программы FastQC.
Часть II: картирование чтений на референсный геном
Картирование чтений
Чтения, уже очищенные в первой части практикума, были картированы на геном участка хромосомы человека с помощью программы
BWA.
Геномы были проиндексированы с помощью комманды:
"bwa index chr13.fasta".
Затем было построено выравнивание прочтений и референса в формате .sam при помощи команды:
"bwa mem chr13.fasta trimm.fastq > out.sam".
Анализ выравнивания
Затем для анализа полученных результатов использовалась программа
SAMtools.
Сначала файл был переведён из формата .sam в .bam (комманда
view с опциями
-b -S -h; -h включает заголовки в выходной файл),
затем отсортировать информацию об откартированных чтениях (комманда
sort),
проиндексировать (комманда
index)
и получить нужную нам статистику (команда
idxstats).
Таким образом, последовательно были произведены следующие комманды:
samtools view -b -S -h out.sam > out.bam
samtools sort out.bam out_sorted
samtools index out_sorted.bam
samtools idxstats out_sorted.bam
|
Чтобы узнать, какое количество чтений откартировалось на 13 хромосому, и использовалась последняя программа (samtools idxstats).
В верхней строчке первое число(113169878) означает длину последовательности, а второе(11932) - количество чтений, которые картируются на 13ую хромосоиму, т. е.
все, кроме одного.

Часть III: Анализ SNP
Поиск SNP и инделей
Однонуклеотидный полиморфизм - отличия последовательности ДНК размером в один нуклеотид в геноме представителей одного вида или
между гомологичными участками гомологичных хромосом. Под инделями подразумеваются делеции (потеря учатка хромосомы),
а инсерции (вставка в нуклеотидной последовательности).
Поиск однонуклеотидных полиморфизмов и инделей, содержащихся в чтениях, очистка и картирование которых была произведена
во время выполнения заданий предыдущих частей практикума. Для поиска однонуклеотидных полиморфизмов и инделей использовались
программы
samtools и
bcftools.
Чтобы создать файл с полиморфизмами в формате .bcf, использовалась программа:
"samtools mpileup -uf chr13.fasta out_sorted.bam -o chr13_snp.bcf".
Опция
-u - несжатый вариант .bcf файла, в то время как
-f говорит о том, что файл с референсной
последовательностью представлен в формате fasta. Файл
chr13_snp.bcf
Затем был получен файл
chr13_snp.vcf с помощью программы bcftools call со
списком отличий картированного генома и референсной последовательности.
В файле
chr13_snp.vcf содержится 137 найденных отличий от референса, среди которых 13 инделей.
| Координата |
Полиморфизм |
Референс (REF) |
Чтения (ALT) |
Глубина покрытия (DP) |
Качество чтений(QUAL) |
| 50092127 |
Индель (делеция) |
TACTCA |
TA |
102 |
217.468 |
| 25511130 |
Инсерция (вставка) |
T |
TGA |
9 |
165.469 |
| 25506914 |
Замена |
G |
T |
3.54462 |
2 |
Аннотация SNP
Полученный список SNP (без инделей) нужно было проаннотировать с помощью программы
Annovar,
используя 5 разных баз данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Для работы с программой annovar из .vcf файла необходимо получить файл, с которым умеет работать эта программа.
Это было сделано с помощью скрипта
convert2annovar.pl, команда:
./convert2annovar.pl -format vcf4 chr13_snp.vcf -outfile 1_chr13.avinput
База данных refgene
Аннотация получена с помощью команды
./annotate_variation.pl -out 0_refgene -build hg19 1_chr13.avinput /nfs/srv/databases/annovar/humandb/
Все sn p делятся на hom и het - их 48 и 59 соответственно. Для экзонных snp выделяют гомологичные и негомологичные замены.
База данных dbsnp
Аннотация получена с помощью команды
./annotate_variation.pl -filter -out 0_dbsn p -build hg19 -dbtype snp138 1_chr13.avinput
/nfs/srv/databases/annovar/humandb/
База данных 1000 genomes
Аннотация получена с помощью команды
./annotate_variation.pl -filter -out 0_1000genomes -build hg19 -dbtype 1000g2014oct_all 1_chr13.avinput /nfs/srv/databases/a
nnovar/humandb/
База данных GWAS
Аннотация получена с помощью команды
./annotate_variation.pl -regionanno -out 0_gwas -build hg19 -dbtype gwasCatalog 1_chr13.avinput
/nfs/srv/databases/annovar/humandb/
База данных Clinvar
Аннотация получена с помощью команды
./annotate_variation.pl -filter -out 0_clinvar -build hg19 -dbtype clinvar_20140211 1_chr13.avin put
/nfs/srv/databases/annovar/humand/
Необходимые для анализа результаты аннотаций по этим базам данных представлены в файле
table.xls (
то же в формате .xml)
Результаты :
В .vcf файле полиморфизмов: 13 инделей и 124 snp.
Однонуклеотидный полиморфизм (Single nucleotide polymorphism, SNP) — отличия последовательности ДНК размером в один нуклеотид (A, T, G или C) в геноме (или в другой
сравниваемой последовательности) представителей одного вида или между гомологичными участками гомологичных хромосом.
В базе данных RefSeq snp делится по категориям exonic, splicing, ncRNA, UTR5, UTR3, intronic, upstream, downstream, intergenic.

В данном случае в exonic попало 7 snp, intronic - 84, 1 downstream и 1 upstream.
Хорошее ли покрытие и качество у найденных полиморфизмов? Итак, 27 SNP имеют покрытие > 10, a 9 - больше 5, то есть довольно хорошее.
Вторая половина имеет покрытие <= 4, причем 40 из них имеют покрытие 1 (соответствующее качество чтений для них тоже низко, часто менее 10, возможно, что это
просто ошибки секвенирования, а не действительно полиморфизмы) SNP с хорошим покрытием также имеют высокое качество чтений.
К каким нуклеотидным и аминокислотным заменам привели snp? Необходимые данные приведены в файле
exonic_variant_function,
полученном при аннотации по refseq. В нем содержится информация о заменах в экзонах генов и их функциональности.
А базы данных GWAS и Clinvar предоставляют клинические аннотации.
По базе данных Clinval не было найдено никакой зависимости между анализируемыми snp и существующими заболеваниями.
По базе данных GWAS три замены ассоциированы со следующими призраками человека:
черты, схожие с ожирением (Obesity-related traits)
сердечная гипертрофия (Cardiac hypertrophy)
артериальная ригидность, жёсткость (Arterial stiffness)
Использованные команды:
| fastqc chr13.fastq |
анализ чтений |
| java -jar /usr/share/java/trimmomatic.jar SE |
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq trimm.fastq TRAILING:20 MINLEN:50 – очистка чтений: с конца удалены нуклеотиды с качеством ниже 20, и все чтения длиной меньше 50 нуклеотидов |
| bwa index chr13.fasta |
индексирование референсной последовательности |
| bwa mem chr13.fasta chr13_tr.fastq > chr13.sam |
построение выравнивания прочтения и референса |
| samtools view -b -o chr13.bam chr13.sam |
перевод файла в бинарный формат .bam |
| samtools sort -T /tmp/chr13_sorted -o chr13_sorted.bam chr13.bam |
сортировка по координате |
| samtools index chr13_sorted.bam |
индексирование |
| samtools idxstats chr13_sorted.bam |
подсчет количества откартированных чтений |
| samtools mpileup -uf chr13.fasta chr13_sorted.bam -o chr13_snp.bcf |
создание файла с полиморфизмами |
| bcftools call -cv chr13_snp.bcf -o chr13_snp.vcf |
создание файла со списком отличий между референсом и чтениями |
| ./convert2annovar.pl -format vcf4 chr13_snp.vcf -outfile 1_chr13.avinput |
создание файла, с которым может работать annovar |
| ./annotate_variation.pl -out 0_refgene -build hg19 1_chr13.avinput /nfs/srv/databases/annovar/humandb/ |
аннотирует файл по базе |
| ./annotate_variation.pl -filter -out 0_dbsnp -build hg19 -dbtype snp138 1_chr13.avinput /nfs/srv/databases/annovar/humandb/ |
аннотирует по базе данных dbsnp |
| ./annotate_variation.pl -filter -out 0_1000genomes -build hg19 -dbtype 1000g2014oct_all 1_chr13.avinput /nfs/srv/databases/annovar/humandb/ |
аннотирует файл по базе данных 1000genomes |
| ./annotate_variation.pl -regionanno -out 0_gwas -build hg19 -dbtype gwasCatalog 1_chr13.avinput /nfs/srv/databases/annovar/humandb/ |
аннотирует файл по базе данных GWAS |
| ./annotate_variation.pl -filter -out 0_clinvar -build hg19 -dbtype clinvar_20140211 1_chr13.avinput /nfs/srv/databases/annovar/humand |
аннотирует файл по базе данных Clinvar |
СПАСИБО ЗА ПРОСМОТР
© Мария Медведева