|
|
1. Поиск структурных гомологов белка 1A4X.
Использовали поиск по сходству структур в PDBeFold.
Поиск осуществлялся по цепи A из PDB-структуры 1A4X с RMSD между 0,8 и 2,5 и длиной выравнивания более 50% от длины 5OWI.
В таблице 1 представлена краткая характеристика найденных гомологов.
|
Таблица 1. Характеристика выбранных структур, гомологичных PurR(1A4X, цепь A),
по результатам поиска в PDBeFold.
|
| PDB |
Организм |
Длина белка (а.о.) |
Длина выравнивания (а.о.) |
RMSD |
| 1UFR |
THERMUS THERMOPHILUS |
167 |
160 (58%) |
0.95 |
| 4p86 |
BACILLUS SUBTILIS |
177 |
164 (100%) |
1.00 |
| 5IAO |
MYCOBACTERIUM TUBERCULOSIS |
171 |
155 (54%) |
1.00 |
| 1W30 |
MYCOBACTERIUM TUBERCULOSIS |
174 |
154 (54%) |
0.87 |
в скобках указан %seq = Nident / Nalign (доля, которую составляет количество идентичных а.о. в выравнивании)
2. Совмещение структур белка и его структурных гомологов
Полученное структурное выравнивание для пяти последовательностей представлено на Рисунке 1.
Скачать выравнивание можно по ссылке.
Результаты структурного выравнивания (файл с совмещёнными сруктурами) предствалены в данном
файле. Это выравнивание представлено на рисунке 2.
Таким образом были совмещены 5 структур. Результат совмещения:
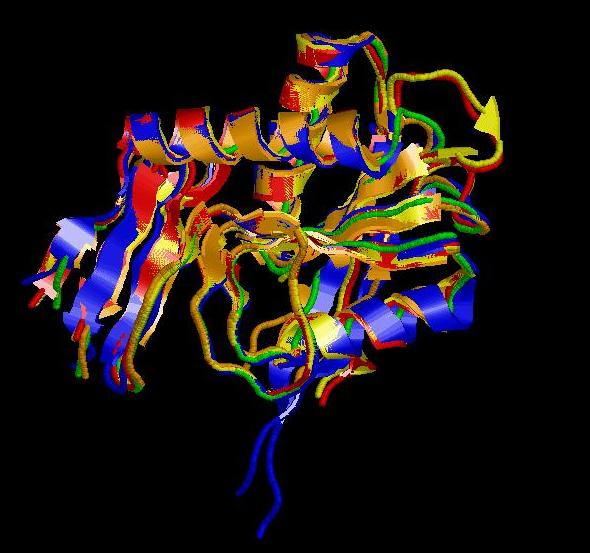 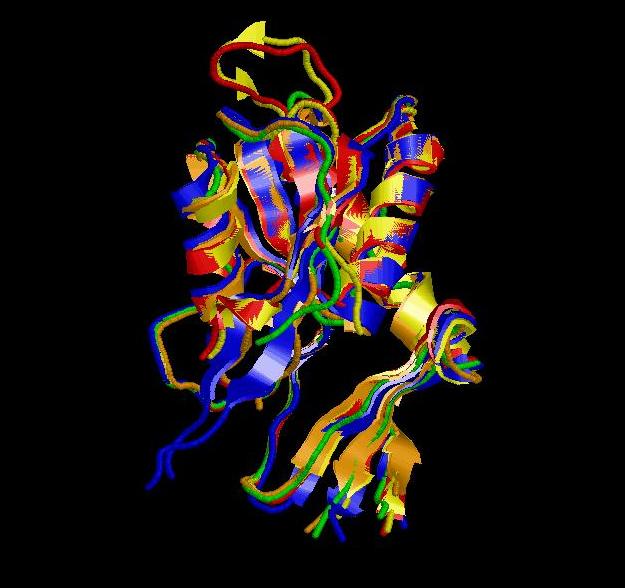
Рисунок 1. Совмещение структур PurR (1A4X, оранжевая цепь) со структурами гомологов из разных организмов.
Видно, что структуры белков достаточно хорошо согласуются и накладываются друг на друга.
Основные отличия наблюдаются в участках петель.
Также было построено выравнивание последовательностей программой Muscle с параметрами по умолчанию.
Это выравнивание представлено на рисунке 3 и доступно по ссылке.
Рисунок 2. Выравнивание по структуре.
Рисунок 3. Выравнивание программой Muscle.
На рисунках 2, 3 видно, что в полученных выравниваниях есть некоторые отличия.
На рисунке 4 представлен один из таких участков выравнивания.
 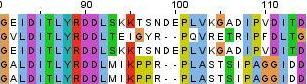
Рисунок 4. Участок с несоответствием выравнивания по структурам (слева) выравниванию по последовательности (справа).
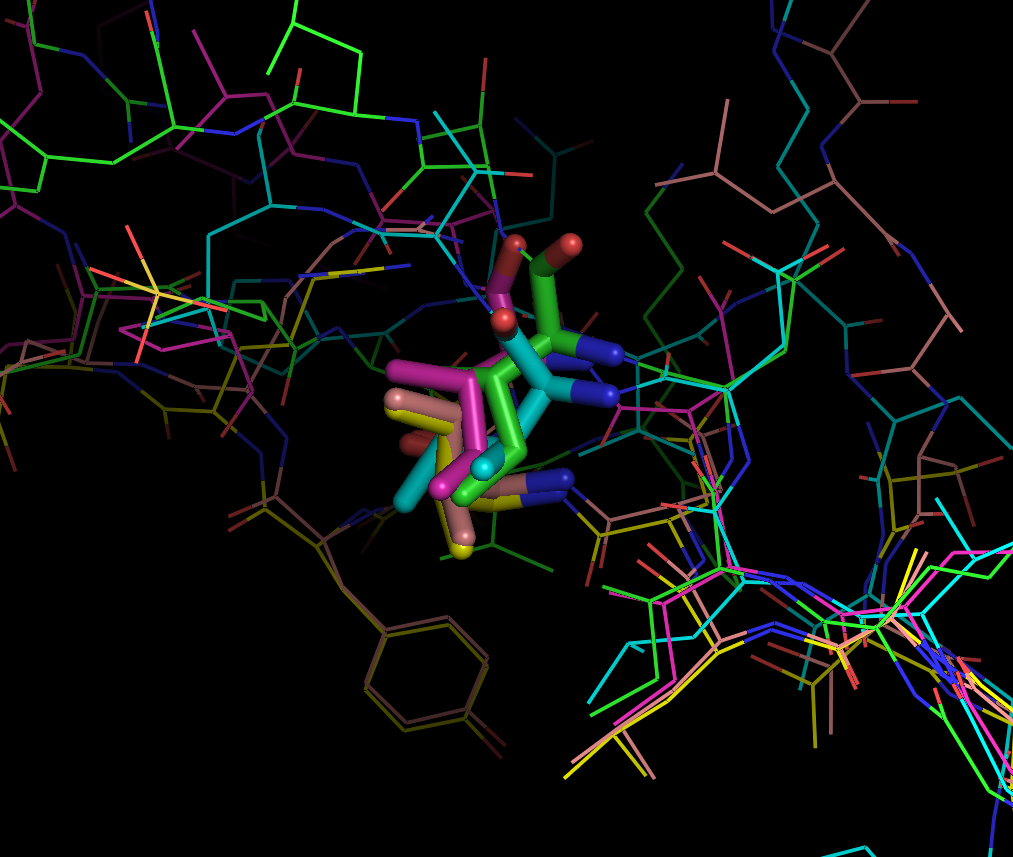
Рисунок 5. Совмещение части структур 1A4X, (жёлтая) со структурами гомологов: 1UFR, 5IAO,
4p86 и 1W30. 76 остатки (85 по выравниванию по последовательности) выделены sticks,
а соседние остатки выделены lines.
3. Совмещение по заданному выравниванию.
Была выбрана одна структура (1OGA, region d:118-202) константного домена T-клеточного рецептора из цепи альфа
и одна (1QRN, region e:119-246) из цепи бета.
Были построены карты бета-листов при помощи
SheeP.
Карта бета-листов альфа цепочки 1OGA:
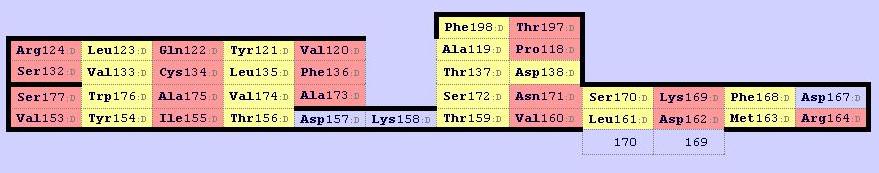
Рисунок 6. Карта бета-листов альфа цепочки 1OGA, полученная с помощью программы SheeP.
Карта бета-листов бета цепочки 1QRN:

Рисунок 7. Карта бета-листов альфа цепочки 1QRN, полученная с помощью программы SheeP.
Для построения совмещения структур были выбраны С-альфа атомы консервативного цистеина (Cys24 для 1OGA и Cys23 для 1QRN)
и окружающих аминокислот.
Команды для построения совмещения:
load 1oga.pdb, 1oga
load 1grn.pdb, 1qrn
select d, 1oga and resi 121-123+133-135+174-176 and name CA and chain d
select e, 1qrn and resi 128-130+146-148+193-195 and name CA and chain e
pair_fit e, d
При выравнивании RMS = 0.505.
Полученное совмещение доменов показано на рисунке 8.
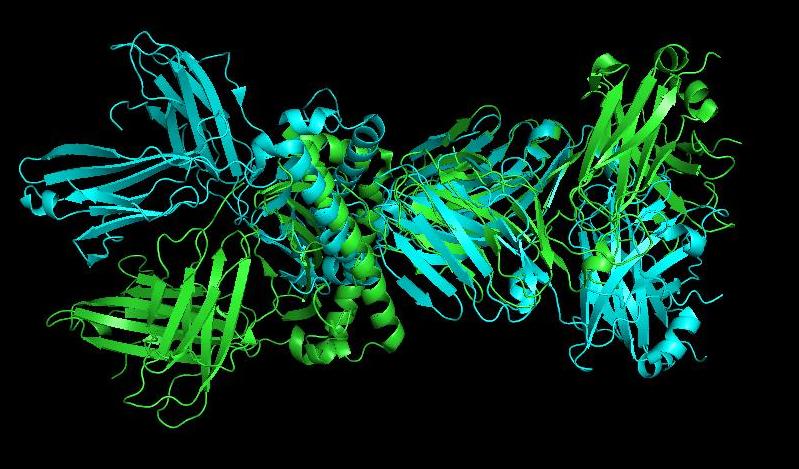
Рисунок 8. Совмещение α-цепи (зелёная) и β-цепи (синяя) в PyMOL.
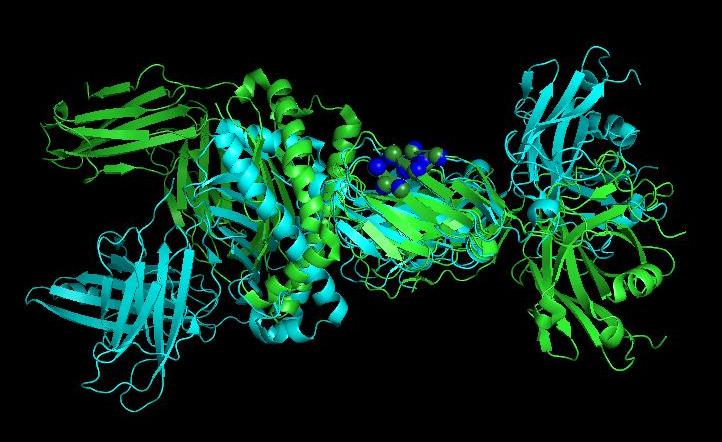
Рисунок 9. Совмещение α-цепи (зелёная) и β-цепи (синяя) в PyMOL.
Шариками показаны атомы, по которым проводилось совмещение, они выделены тёмно-зелёным и тёмно-синим цветами соответственно.
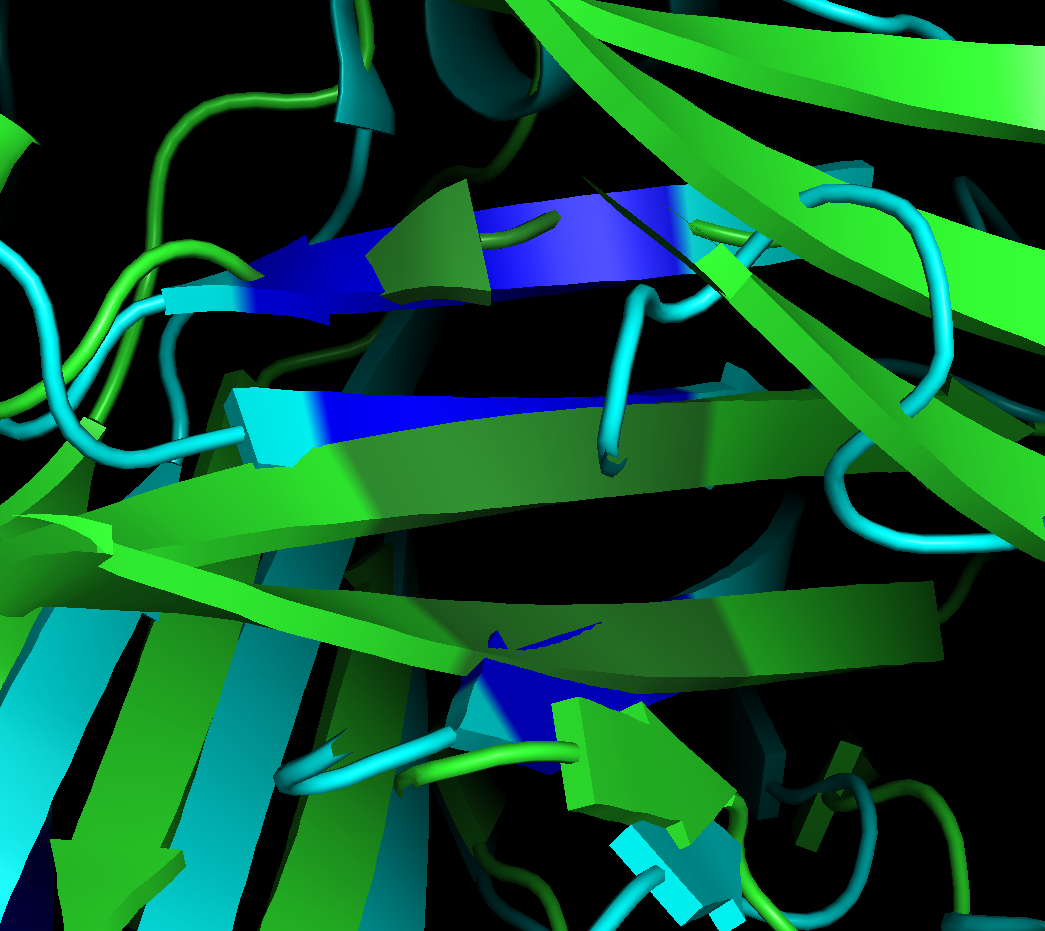
Рисунок 10. Совмещение α-цепи (зелёная) и β-цепи (синяя) в PyMOL.
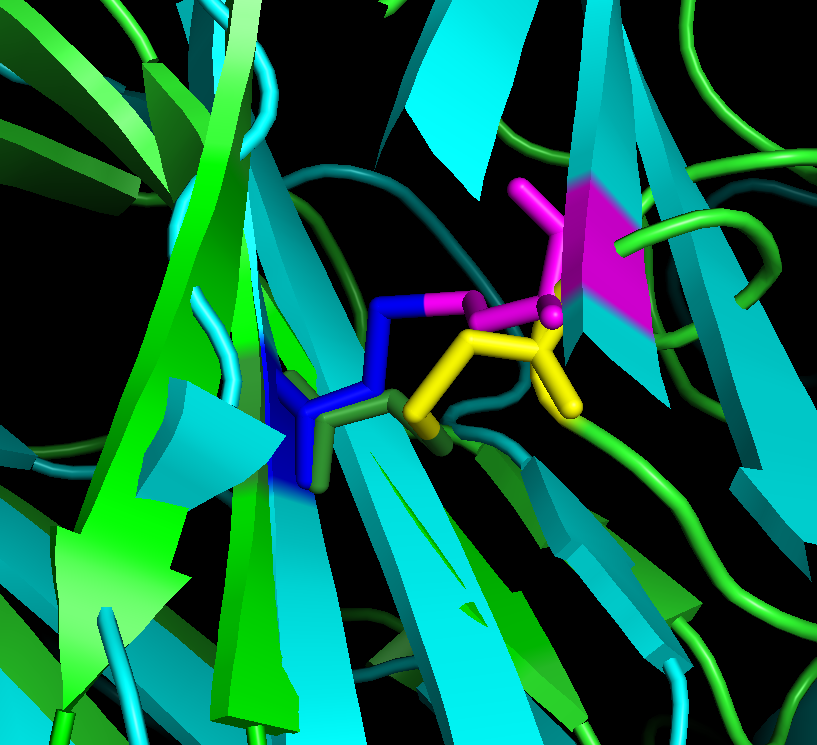
Рисунок 11. Совмещение α-цепи (зелёная) и β-цепи (синяя) в PyMOL. Показаны
цистеины, образующие дисульфидные связи: синим и фиолетовым (из1qrn), зелёным и жёлтым (из 1oga).
По рисункам выше видно, что, в целом, совмещение не очень хорошее, при приближении нужные участки тоже выровнены
не очень. Однако видно, что топологии структур схожи (если "пройтись глазами" по остовам цепей).
|


