Валидация
Задание 1.
Этот практикум посвящен анализу структуры SARS-CoV nsp14-nsp10 комплекса с функциональным лигандом SAM (PDB ID: 5C8T), чтобы определить ее пригодность для работы по изучению структурных и функциональных свойств.
Коронавирусы принадлежат к семейству Coronaviridae порядка Nidovirales. Они представляют одну из основных угроз для здоровья населения, вызывая острые респираторные заболевания (SARS и MERS). Сейчас это все знают, однако эта информация из статьи 2015 года, которая была написана в рамках получения и исследования данной структуры. Кроме того, там сказано, что к 25 мая 2015 года было зафиксировано 8000 заражений и 800 смертей от SARS-CoV и 1139 заражений и 431 смерть от MERS-CoV.
Неструктурный белок 14 (nsp14) коронавирусов необходим для вирусной репликации и транскрипции. С N-конца это белок содержит экзорибонуклеазный домен (ExoN). Он отвечает за пруфридинг. С С-конца тоже есть домен, он участвует в кэпировании мРНК. Для активации экзорибонуклеазного домена нужен неструктурный белок 10 (nsp10). Nsp10 взаимодействует с ExoN доменом, стабилизируя его и стимулируя его активность.
Оба домена белка nsp14 играют важную роль в вирусной репликации и транскрипции. Исследование структуры комплекса nsp14-nsp10 позволит лучше понять функцию nsp14. В дальнейшем это может быть полезно для разработки антивирусных препаратов.
Чтобы оценить качество структуры, я сначала ознакомилась с отчетом о валидации с pdb.
Далее, описывая характеристики структуры, я буду использовать данные, предоставленные депозиторами.
Разрешение данной структуры 3.20 Å при полноте данных 98.6%.
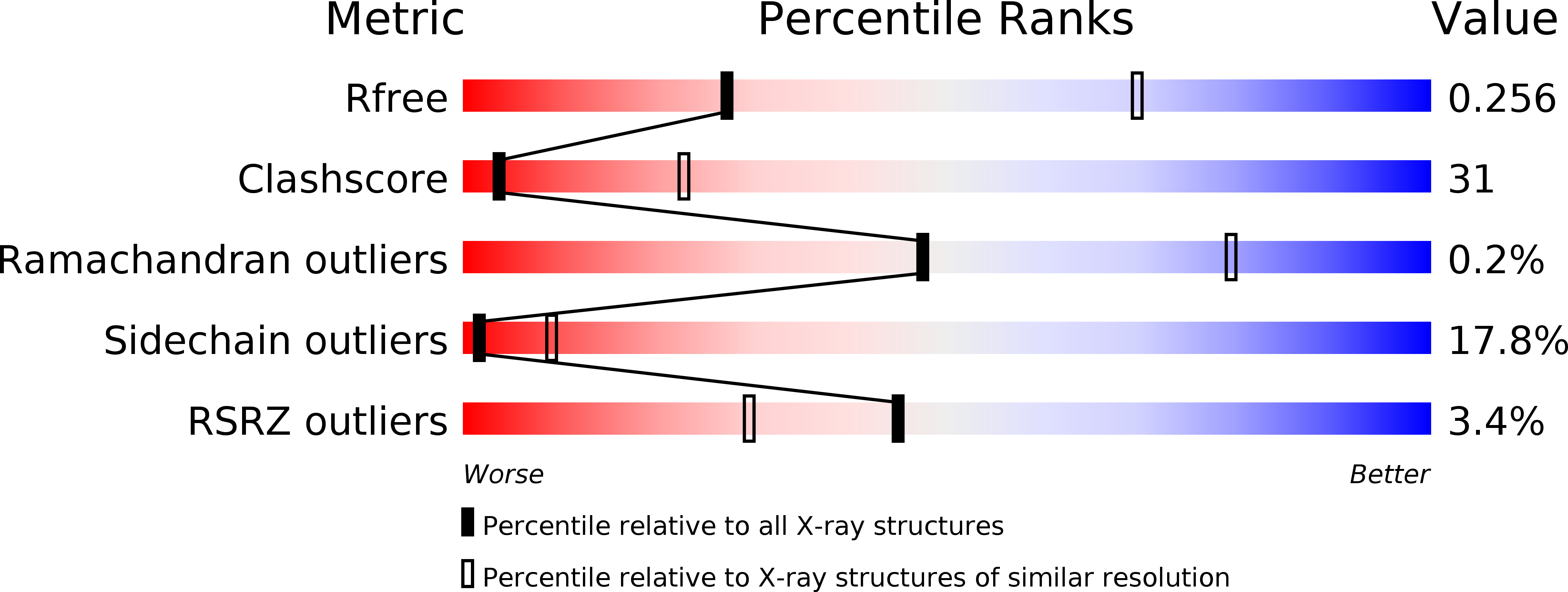
Рисунок 1.
На рисунке 1 изображен график с процентильными баллами (в диапазоне от 0 до 100) для глобальных показателей валидации записи.
R-фактор модели 0.243, R-free при этом равен 0.265. Из рисунка 1 видно, что это неплохой показатель по сравнению со структурами того же разрешения, но по сравнению со всеми структурами, полученными РСА, значение R-free ниже среднего. Однако, по крайней мере, данная модель не переоптимизирована.
Clashscore на 1000 атомов в данной модели равен 31. Что достаточно плохо и в сравнении со структурами того же разрешения, и всеми структурами, полученными РСА. Всего на 10266 атомов не водородов и 9844 атомов добавленных водородов в структуре 627 столкновений. В силу объема данных, я не смогла просмотреть все столкновения, однако, там присутствуют столкновения между добавленными математически водородами, их чего следует, что, вероятно, некоторые столкновения можно не учитывать.
В данной модели всего 3 маргинала по картам Рамачандрана из 1282 проверенных остатков. Это нормальное количество для все структур РСА, а для структур с таким же разрешением - даже хорошее. Все три маргинала находятся в одной цепи в половине димера, в другой половине их нет. Это странно, потому что по идее половины должны быть идентичны. Возможно, это можно объяснить изменениями при кристаллизации, либо какими-то недочетами при обсчете модели.
Маргиналов по ротамерам боковых радикалов остатков в данной модели 199 из 1116 проверенных аминокислотных остатков. Это плохой показатель как для всех структур РСА, так и для структур с таким же разрешением. Они представлены примерно в одном процентном соотношении для всех цепей. Также нет какой-то закономерности по расположению их относительно концов цепей. Они встречаются на всей протяженности цепей. Возможно, они торчат из молекулы и их количество объясняется подвижностью и не очень высоким разрешением, но визуализировать 199 остатков я не стала, поэтому это только гипотеза.
Маргиналов по RSRZ в модели 44 из 1294 проверенных остатков. Для данного разрешения это плоховато, для все структур РСА это почти средний показатель. Маргиналы присутствуют во всех цепях. Такие цифры свидетельствуют о том, что полученная модель не очень хорошо соответствует полученным в результате эксперимента данным. Тем не менее 3.4% маргиналов по RSRZ это не очень много, по моему мнению.
В структуре есть участки, для которых нет электронной плотности. Это С-концы всех цепей. На N-концах модель плохо соответствует электронной плотности. Поскольку это только концы белка и судя по информации в Uniprot, эти участки не несут функциональной значимости.
В целом, по данным из отчета о валидации я могу сказать, что структура не очень хорошая. Много маргинальных остатков, относительно большой R-free, много столкновений.
Задание 2.
В данном задании я более подробно рассмотрела некоторые маргинальные остатки. Кроме отчета о валидации pdb, я пользовалась в этом задании сервисами MolProbity и CheckMyMetal.
Выдача MolProbity по количеству маргиналов практически не отличалась от отчета о валидации pdb. Анализ геометрии всеми доступными контактами после добавления водородов и поворотов Asn/Gln/His (только для остатка цепи D 373 была очевидная необходимость поворота, для еще трех была рекомендация поворота и еще для 18 были возможны оба положения, но поворот предпочтителен) представлен на рисунке 2.
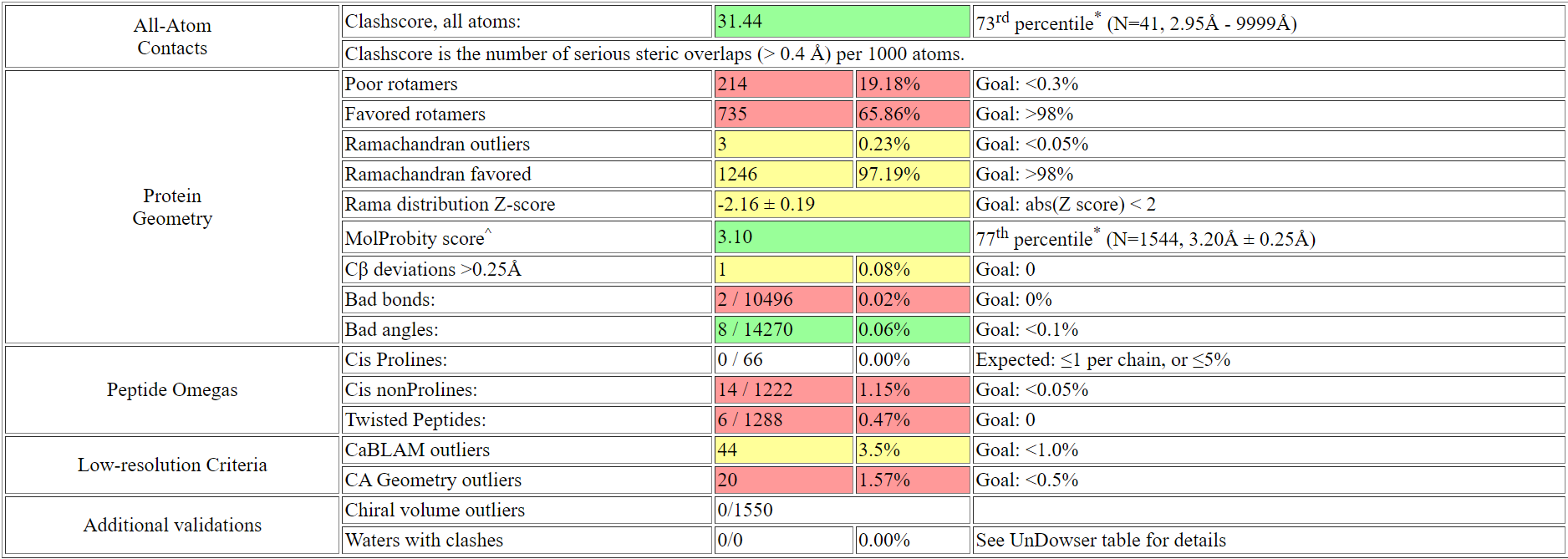
Рисунок 2.
Опираясь на выдачу MolProbity и отчет о валидации я выбрала несколько маргинальных остатков, для которых я сделала картинки, чтобы продемонстрировать их дефекты в модели.
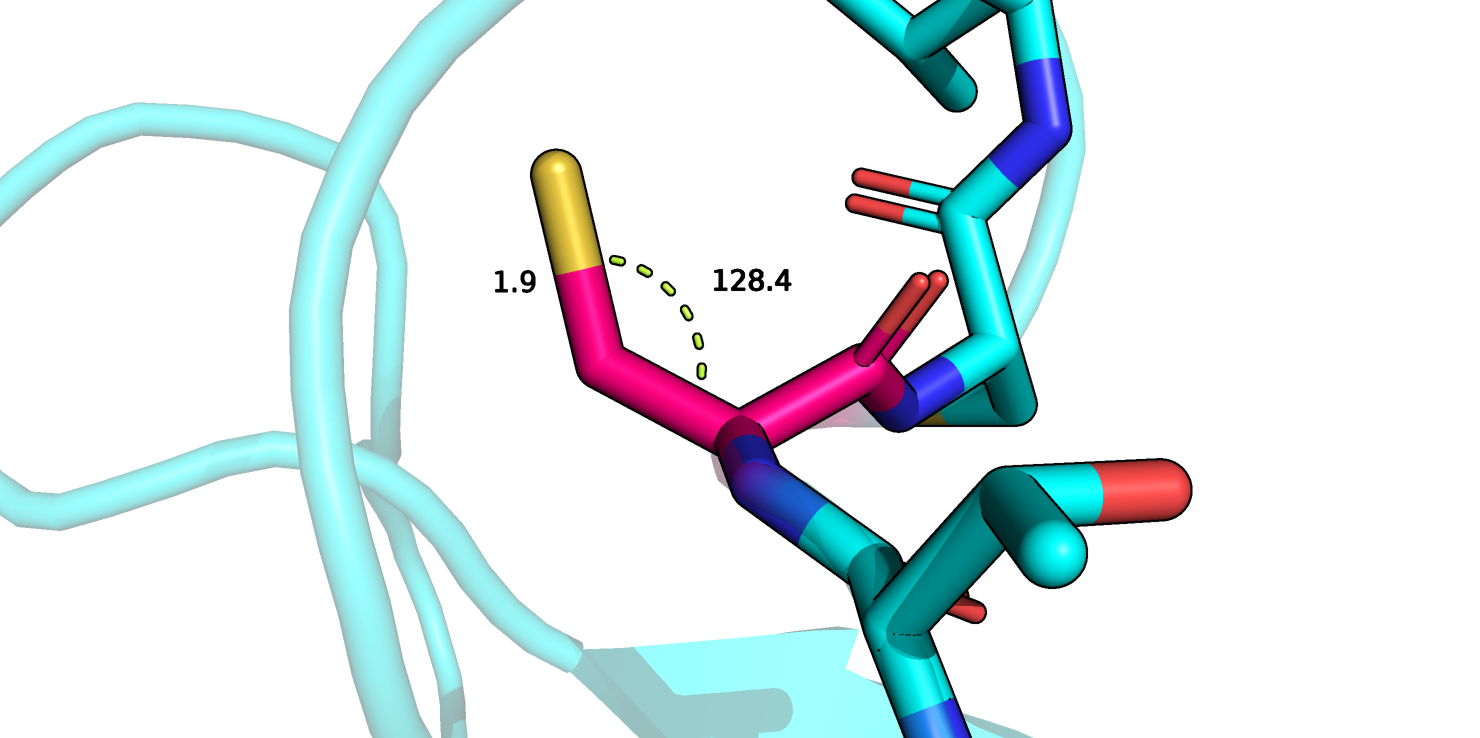
Рисунок 3.
На рисунке 3 изображен 207 цистеин цепи В. Он является маргиналом, потому что длина связи CB-SG, входящей в его состав, составляет 1.93 Å, в то время как идеальная длина связи 1.82 Å (согласно отчету pdb; Z-score = 6.54). Кроме того, угол между атомами CA-CB-SG равен 128,4° при идеальном угле 114° (в отчете pdb Z-score = 8). Также в отчете pdb указано два клэша с участием данного остатка, однако оба клэша с водородами, а поскольку водороды рассчитаны математически, значимость данных клэшей весьма спорная. Согласно анализу MolProbity данный остаток не является маргиналом по длине связи, но является маргиналом по данному углу и образует один клэш с водородом B 226 Cys (этот клэш также есть в отчете pdb). Угол и связь отображены на рисунке 3.
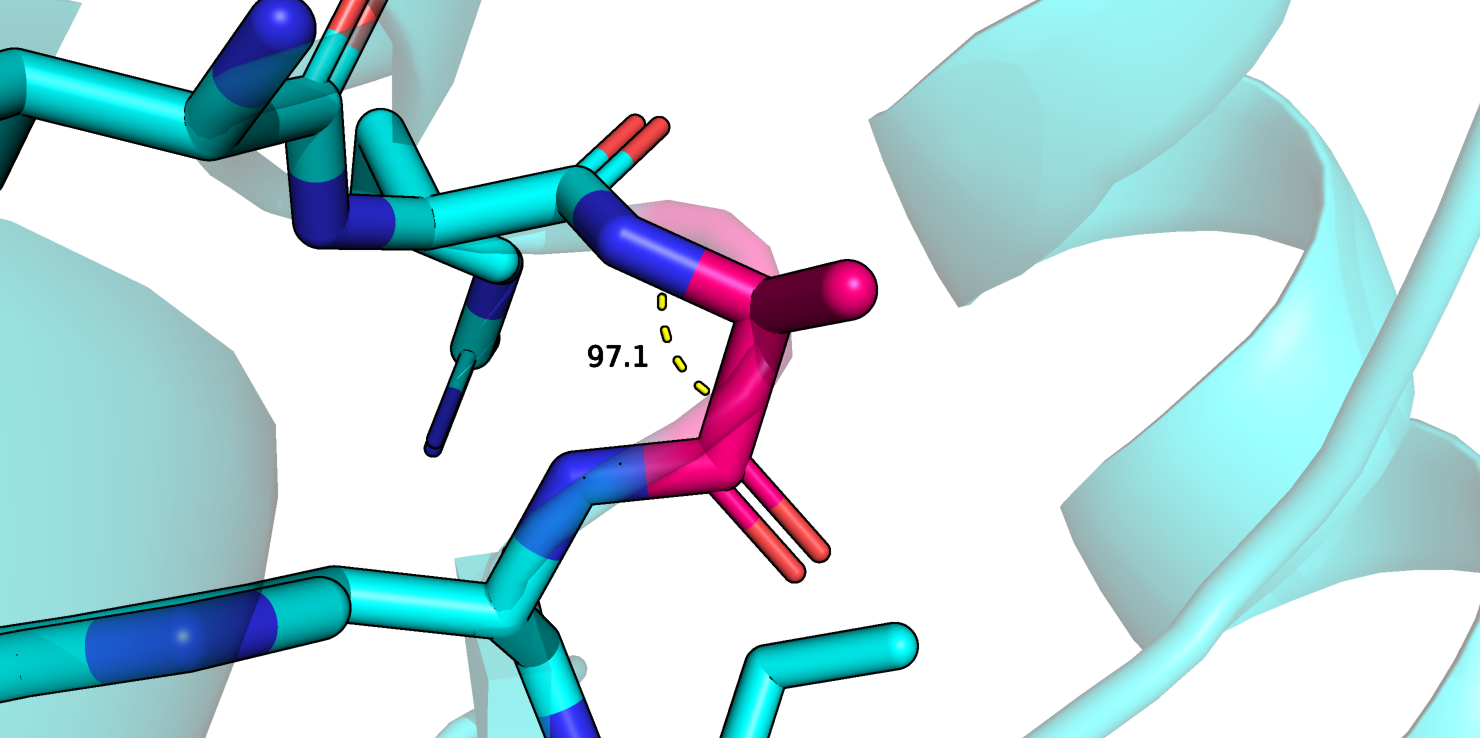
Рисунок 4.
Далее я рассмотрела остаток цепи В 85 Ala, который является маргиналом по величине угла N-CA-C, которая равняется 97.13°, при идеальной величине 111° (согласно отчету pdb Z-score = -5.14). Кроме того в отчете о валидации с pdb этот остаток указан как RSRZ маргинал (RSRZ = 2.2), кроме того, он участвует в двух кэшах с водородами других остатков. Анализ в MolProbity характеризует данный остаток как маргинал по тому же углу, по геометрии атома СА и по углу ω = 139.58. Также он участвует в одном клэше, где его атом водорода сталкивается с атомом углерода B 182 Val (совпадает с клэшем в отчете pdb). Сам остаток и некорректный угол отображены на рисунке 4.
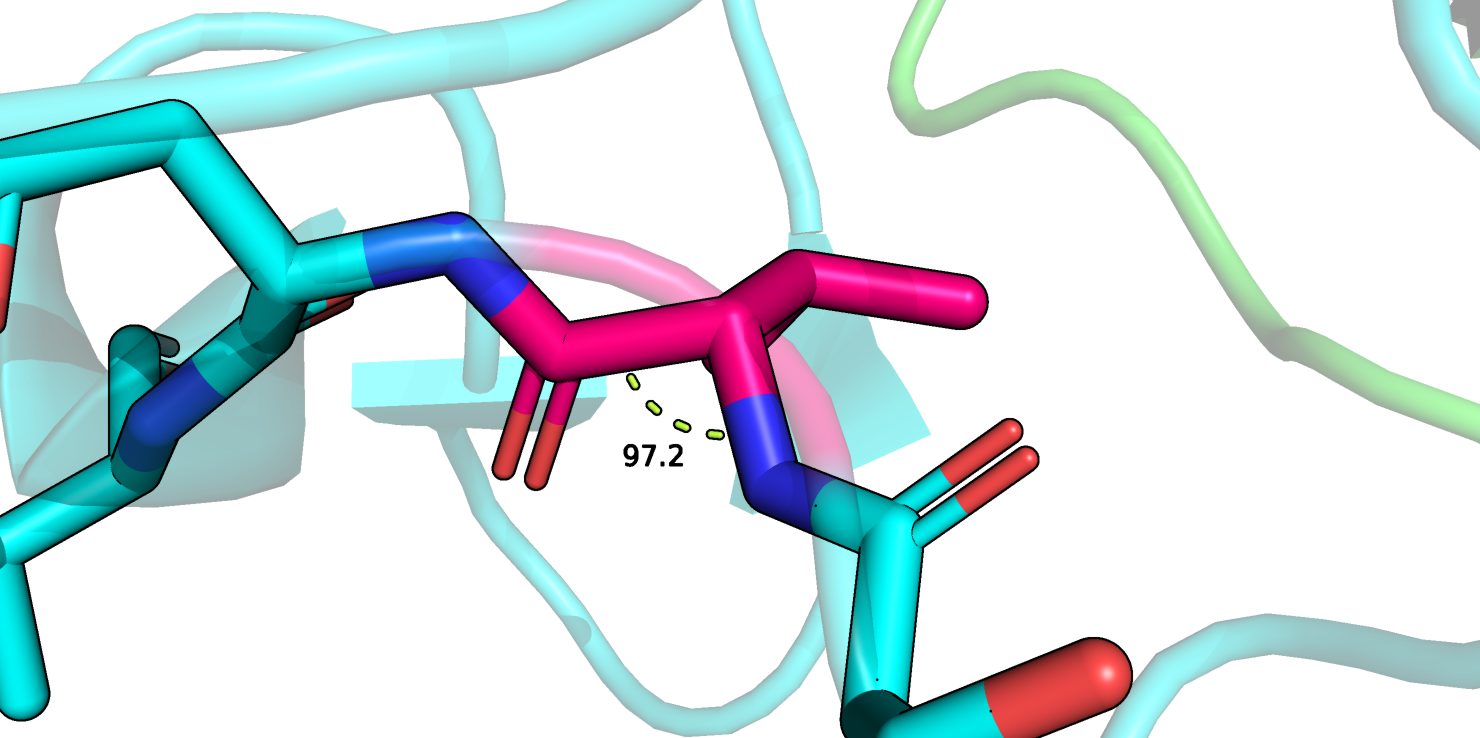
Рисунок 5.
Следующий рассмотренный мной остаток тоже маргинален по углу. Это остаток цепи B 29 Val (рисунок 5). Угол N-CA-C в модели равен 97.22° при идеальном угле 111° (Z-score = -5.1 в отчете о валидации pdb). Также согласно отчету pdb данный остаток образует 3 клэша. Клэши опять с водородами. MolProbity тоже определил этот остаток как маргинал по углу N-CA-C, маргинал по геометрии СА атома и участник клэша с водородом остатка цепи B 34 Lys.
После рассмотрения трех аминокислотных остатков, я решила оценить качество лиганда. Так как предоставленная структура является димером, я решила рассмотреть лиганд цепи В.
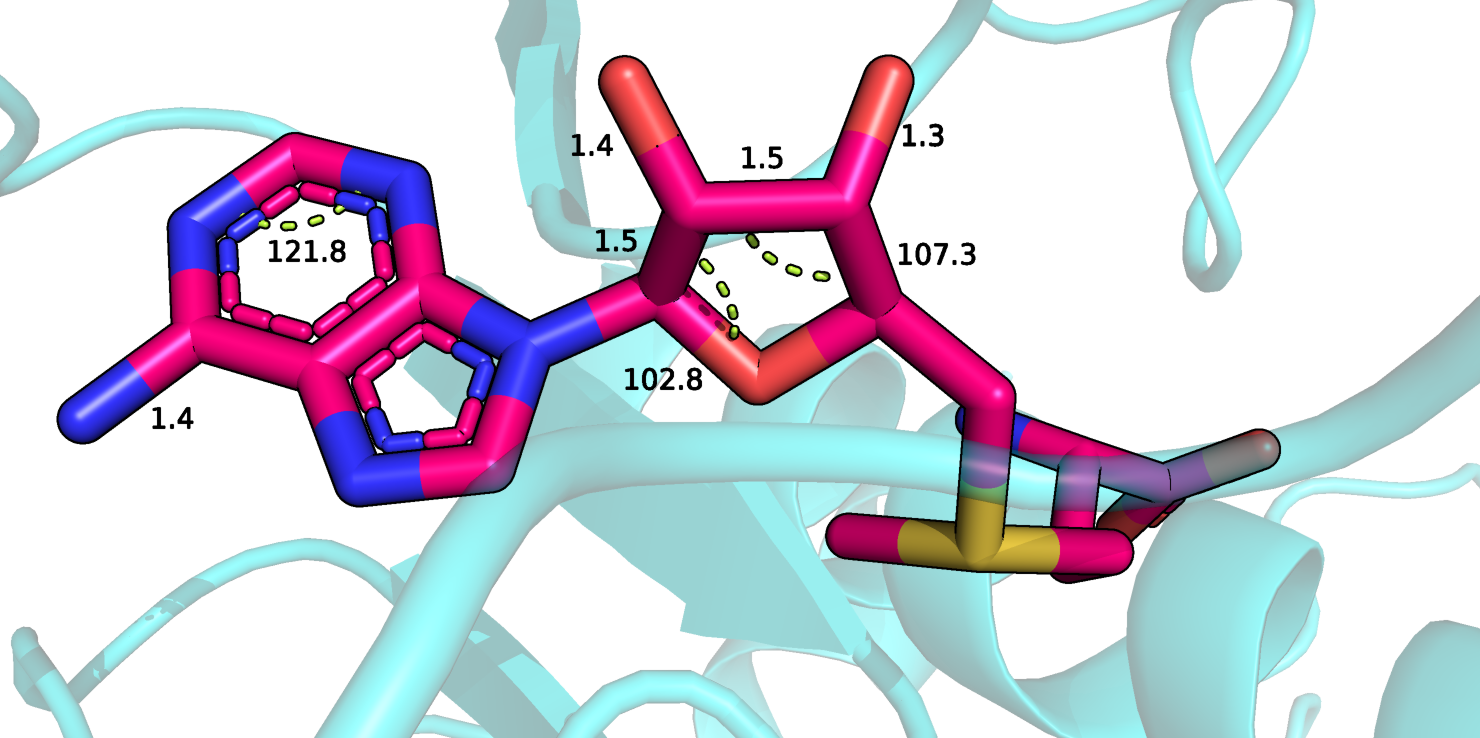
Рисунок 6.
На рисунке 6 изображен лиганд цепи В SAM и подписаны все маргинальные связи и углы. Всего в данном лиганде 3 маргинальных угла, 5 маргинальных связей и 7 торсионных маргиналов. Кроме того лиганд цепи В образует 8 клэшей. Все маргиналы лиганда представлены в таблице 1.
| Тип | Атомы | Расстояние/угол в модели | Идеальное/ый расстояние/угол | Z-score |
|---|---|---|---|---|
| длина связи | O3'-C3' | 1.35 Å | 1.43 Å | -3.39 |
| длина связи | C2'-C1' | 1.49 Å | 1.53 Å | -2.61 |
| длина связи | C6-N6 | 1.43 Å | 1.34 Å | 2.48 |
| длина связи | O2'-C2' | 1.38 Å | 1.43 Å | -2.08 |
| длина связи | C2'-C3' | 1.47 Å | 1.53 Å | -2.04 |
| угол | N3-C2-N1 | 121.78° | 128.68° | -4.41 |
| угол | O4'-C1'-C2' | 102.82° | 106.93° | -2.81 |
| угол | C2'-C3'-C4' | 107.27° | 102.64° | 2.38 |
В целом качество лиганда оставляет желать лучшего.
Данная структура также содержит ионы. Я воспользовалась сервисом CheckMyMetal, чтобы проверить геометрию ионов. Выдача данного сервиса приведена на рисунке 7.
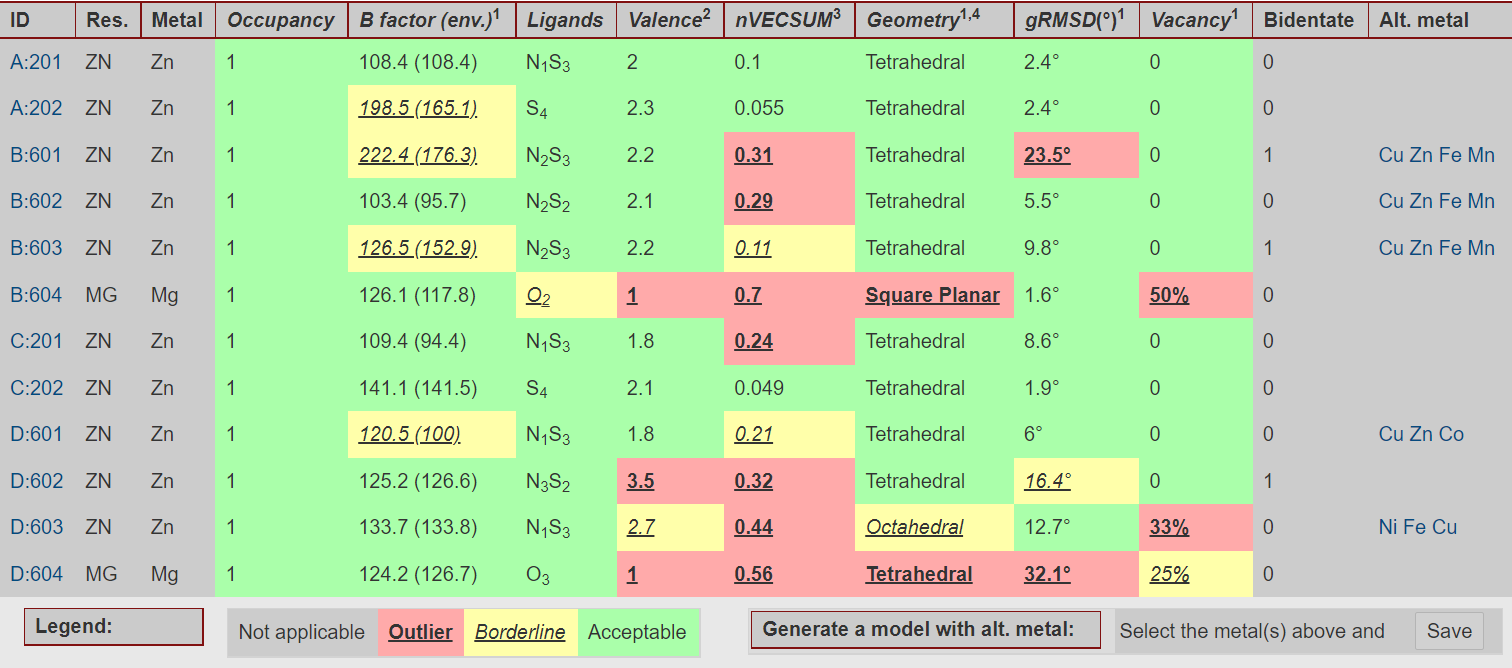
Рисунок 7.
Суммарно получилось, что среди ионов 7 маргиналов. Более подробно я решила рассмотреть ион магния цепи В, потому что у него одни из самых низких показателей качества.
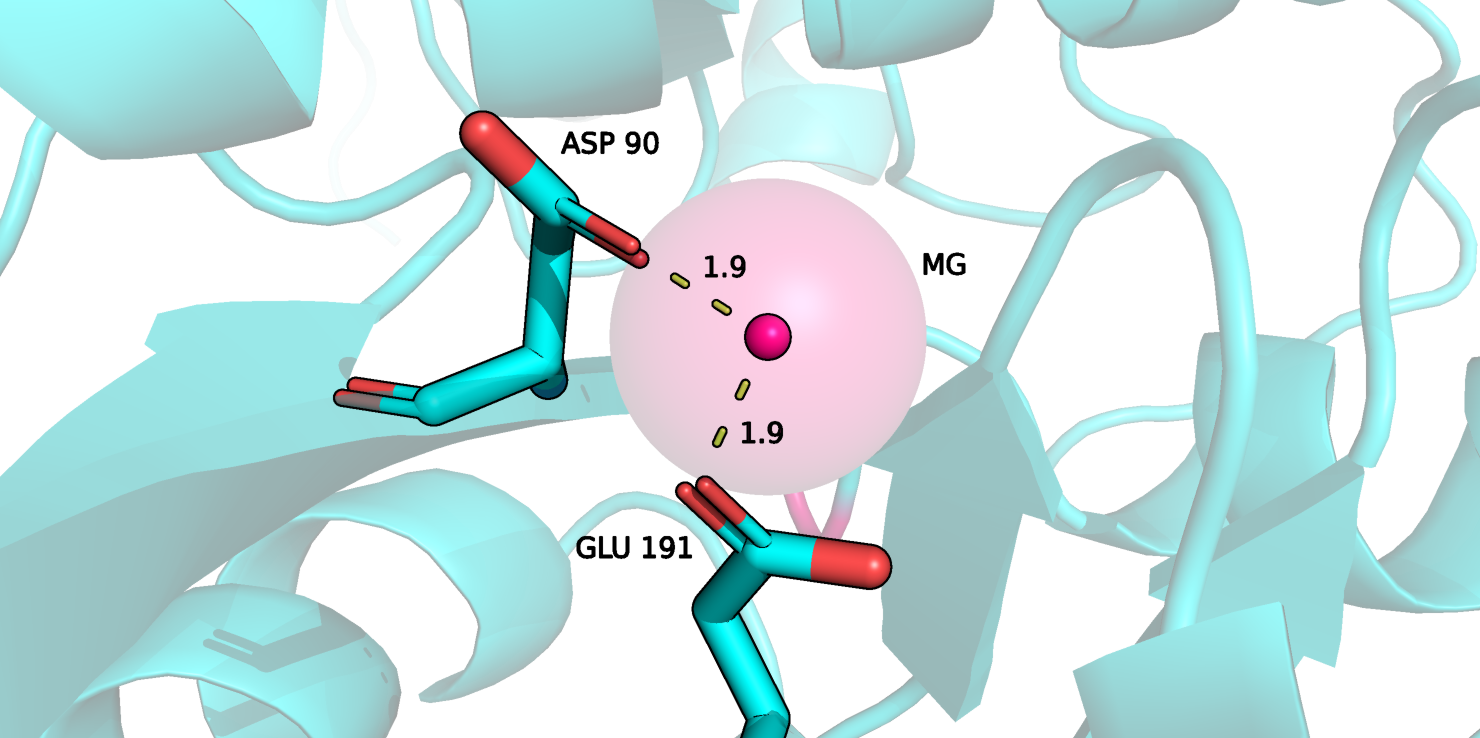
Рисунок 8.
На рисунке изображен атом магния в цепи В и взаимодействующие с ним аминокислотные остатки. Из отчета CheckMyMetal видно, что Mg должен взаимодействовать с тремя атомами, а не с двумя. Показатель симметричности сферы высокий, что говорит о плохой симметричности. Геометрия неправильная, 50% координационных сайтов сферы не заняты.
Задание 3.
В целом качество структуры оставляет желать лучшего. Очень много маргиналов. Симметричные структуры отличаются друг от друга. Много клэшей. Электронная плотность покрывает почти всю молекулу, однако с разрешением в 3.2 Å атомы плохо различимы, и сложно определить точное их положение. Некоторые аминокислотные остатки, находящиеся по краям молекулы не покрыты электронной плотностью. Как уже было написано выше, лиганд важен для функций данных белков, однако качество его структуры тоже плохое, при том, что лиганд находится внутри молекулы и полностью покрыт электронной плотностью. Поскольку маргинальные остатки встречаются во всех местах молекулы, они присутствуют и в функционально важных участках. Хоть в исходной статье и изучаются биологические функции на основе данной структуры, я считаю, что структура слишком низкого качества для того, чтобы делать выводы о каких-то функциях.
Задание 4.
Кроме модели с PDB, есть еще переделанная модель с PDB Redo. Но её качественность тоже подлежит сомнению. Поэтому в данном задании я сравниваю две модели.
С точки зрения вписанности в электронную плотность особо ничего не изменилось. Также покрыта наибольшая часть молекулы, но есть торчащие аминокислотные остатки на периферии.
Для переделанной модели я тоже сделала анализ на MolProbity. Выдачу данного сервиса после анализа полной геометрии структуры с добавленными водородами можно увидеть на рисунке 9.
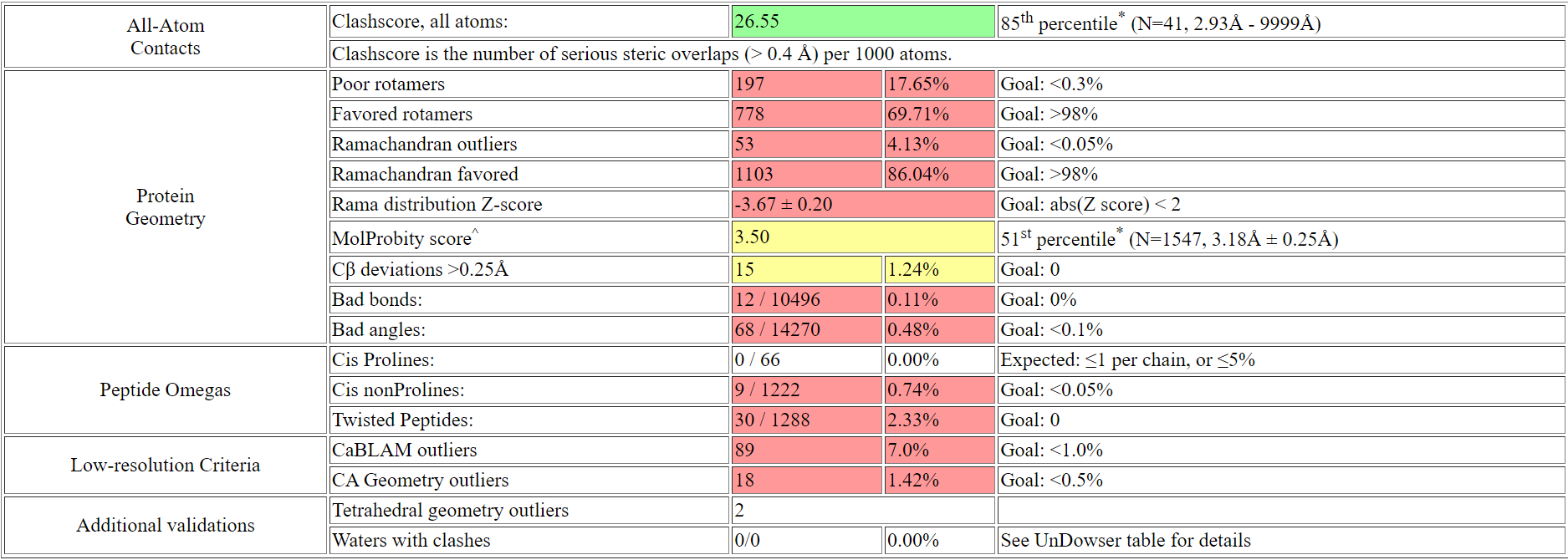
Рисунок 9.
Для переделанной структуры увеличилось количество маргиналов по рамачандрану, по величине углов, по длине связей и вообще в выдаче стало больше красных ячеек в табличке. Тем не менее, clashscore стал немного ниже и стало немного меньше маргиналов по ротамерам боковых радикалов остатков. Но в целом показатели упали, что отлично видно при сравнении рисунков 2 и 9.
Потом я проверила улучшилось ли качество отдельных участков молекулы, рассмотренных в задании 2. Далее на рисунках розовый остаток принадлежит структуре PDB, а зеленый - PDB Redo.
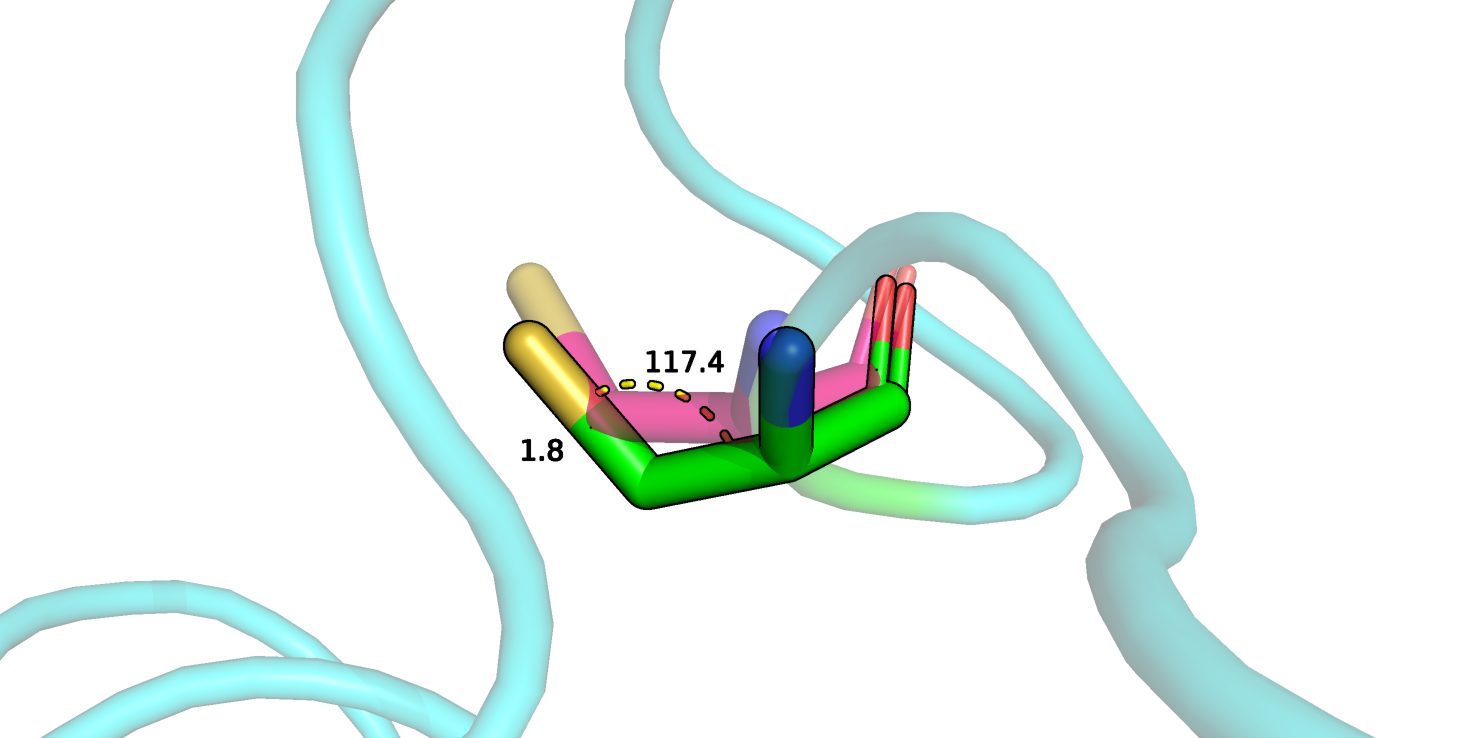
Рисунок 10.
Остаток цистеина (рисунок 10) стал выглядеть значительно лучше, длина связи (1.8 Å) уже больше не маргинальна, угол тоже стал ближе к идеальному: вместо 128.4° он стал равен 117.4° (идеальный 114°).
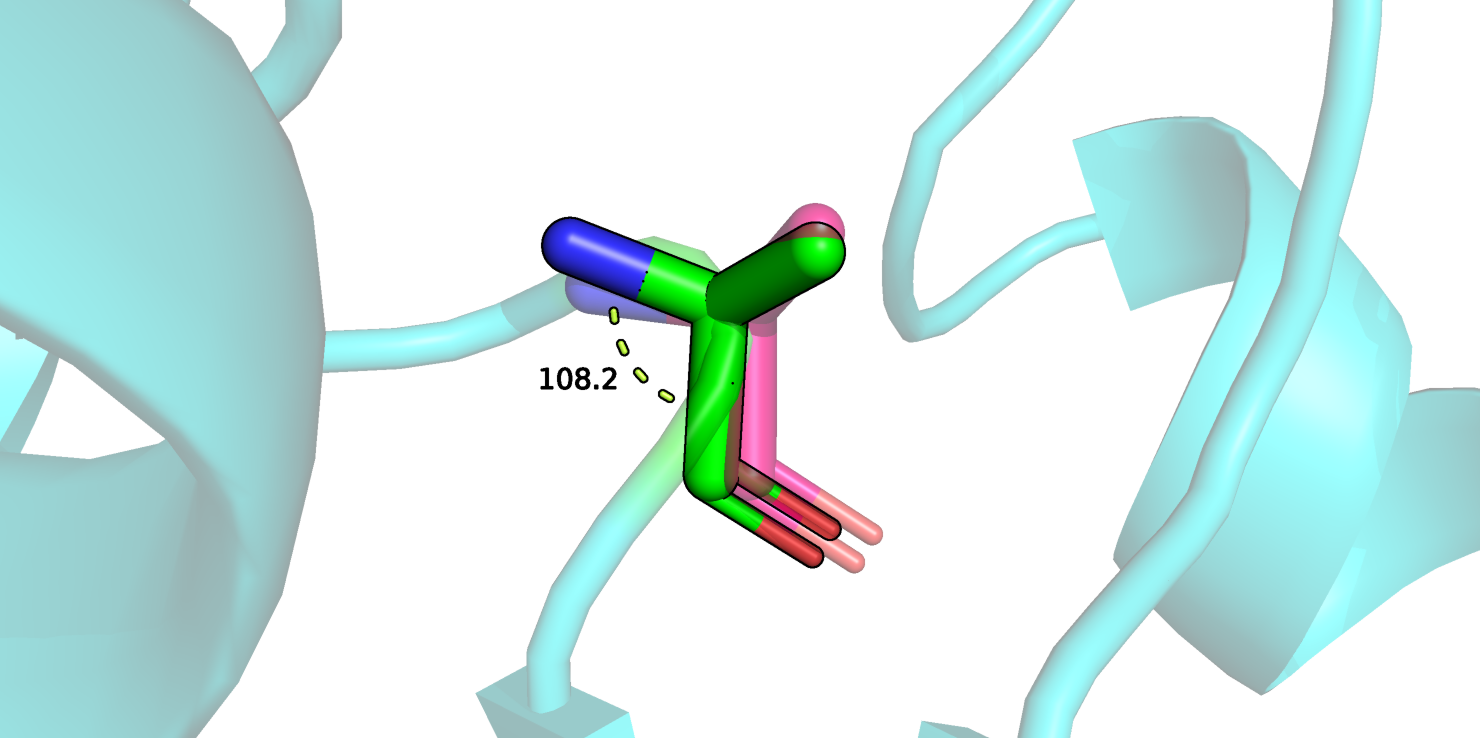
Рисунок 11.
На рисунке 11 изображен остаток аланина, который тоже перестал быть маргинальным по величине угла. Угол стал 108.2° при идеальном 111°.
Рисунок 12.
Угол в остатке валина значительно приблизился к идеальному. Вместо 97.2° он стал 105.2° при идеальном 111°. Этот остаток можно увидеть на рисунке 12.
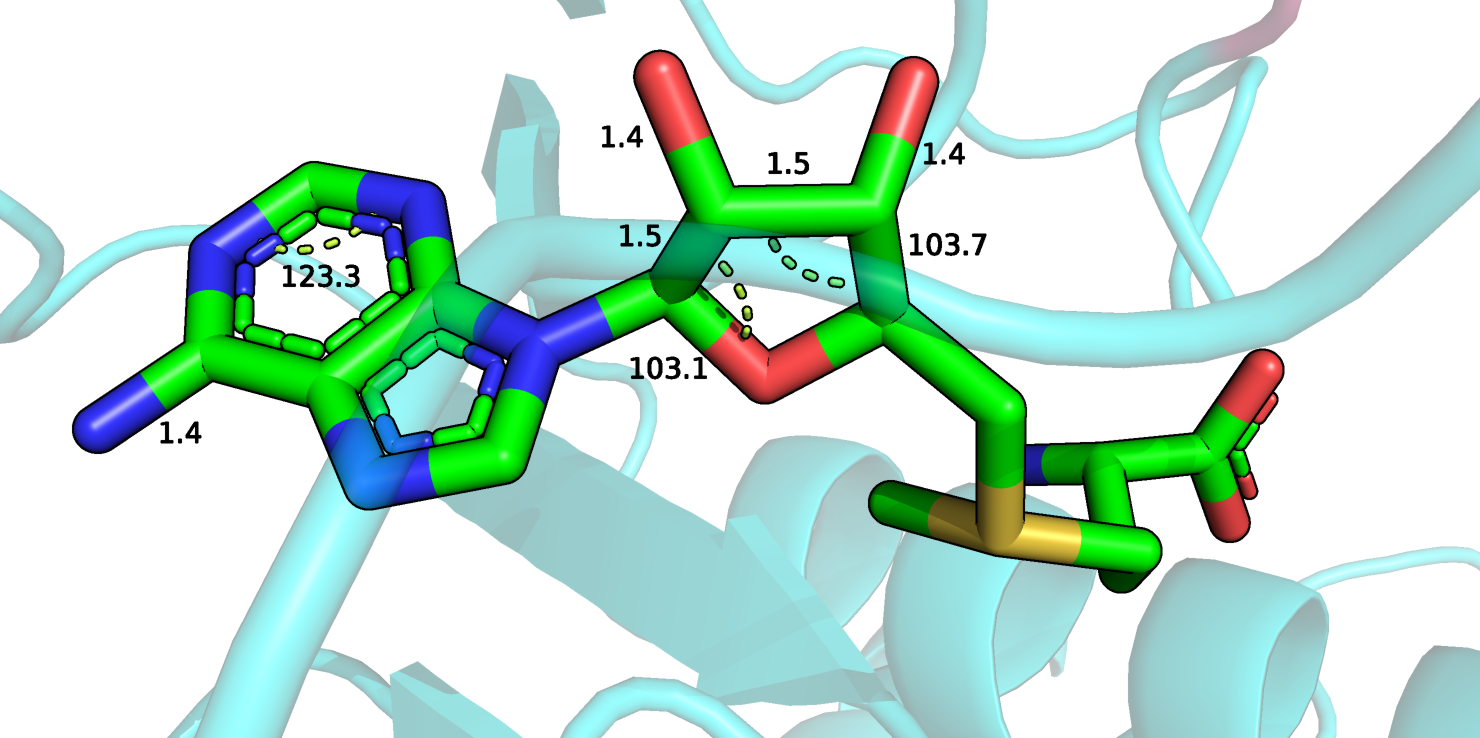
Рисунок 13.
Для лиганда SAM показатели тоже улучшились, но далеко не все. Сам лиганд и значения углов и длин связей можно увидеть на рисунке 13. Для большинства длин связей ничего не изменилось, однако все углы немного изменили свои значения в сторону идеальных. Тем не менее качество модели лиганда осталось плохим.
Модель PDB Redo я проверила и на сервисе CheckMyMetal. Выдачу этого сервиса можно увидеть на рисунке 14.
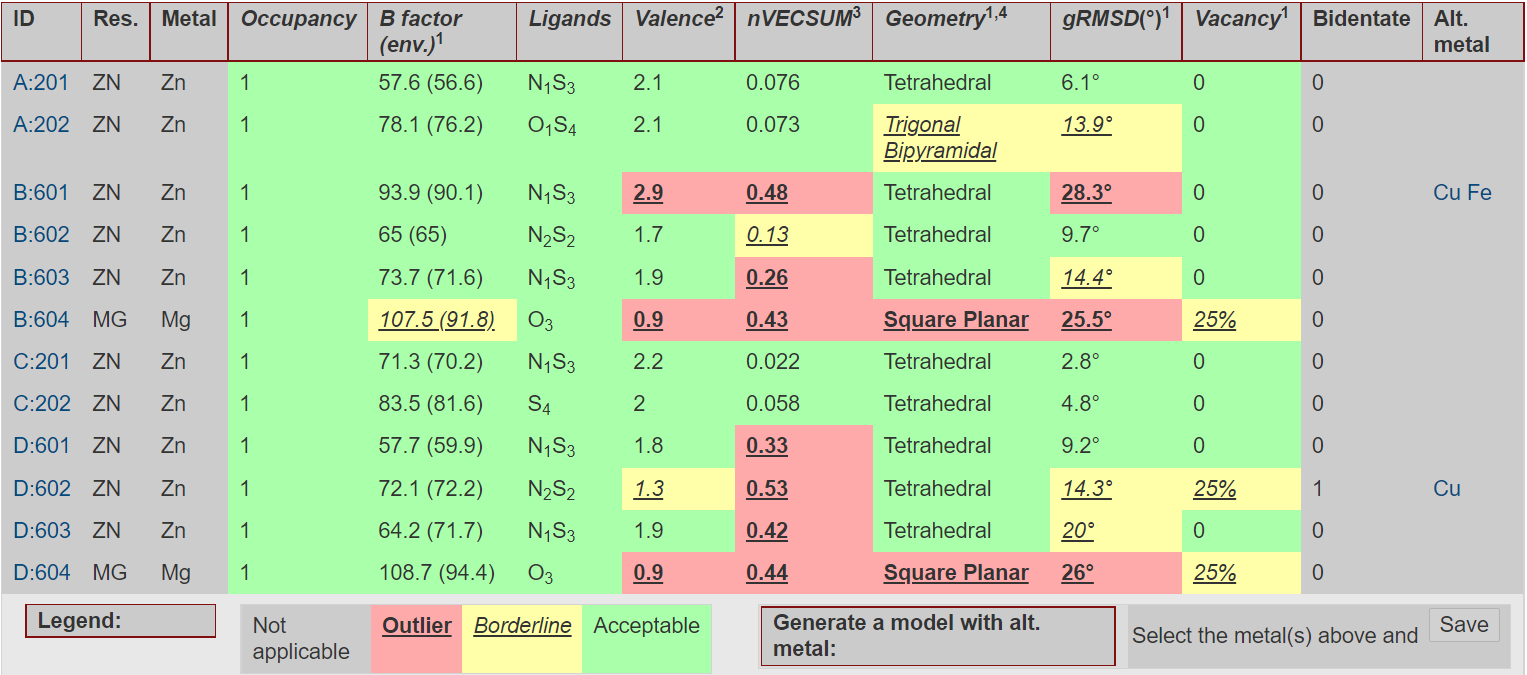
Рисунок 14.
Результат анализа с помощью данного сервиса показал более хорошие результаты для переделанной структуры, чем для структуры PDB. Но маргинальных ионов осталось 7. Просто некоторые показатели заметно улучшились.
Что касается иона магния, рассмотренного мной в задании 2, то для него стал нормальным показатель Ligands. Улучшились показатели валентности, симметричности и вакантных сайтов. Однако геометрия осталась неправильной.
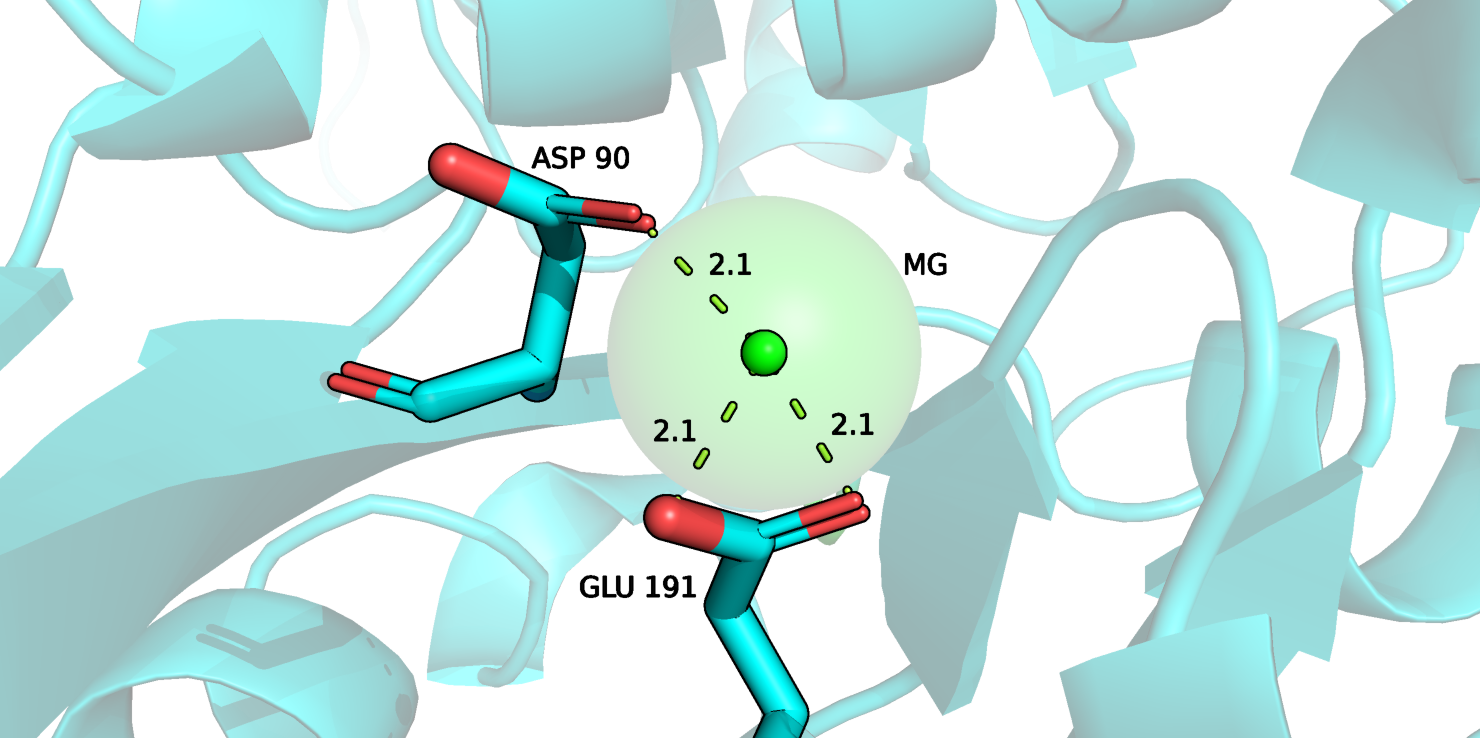
Рисунок 15.
На рисунке 15 изображен ион магния и взаимодействующие с ним аминокислотные остатки в переделанной структуре PDB Redo. Как видно, теперь атом магния координирован тремя кислородами, а не двумя как в структуре PDB.
Не смотря на то, что показатели для рассмотренных мной частей модели улучшились, они во многом далеки от хороших. В целом качество структуры PDB Redo плохое, по некоторым параметрам даже хуже, чем структуры PDB. Я считаю, что эту структуру тоже нельзя использовать для изучения свойств белка.