Исследование белок-кодирующих последовательностей генома Achromobacter xylosoxidans
Аннотация
В ходе настоящего исследования были изучены распределения длины белков и размера перекрывающихся участков белок-кодирующих последовательностей бактерии Achromobacter xylosoxidans. Помимо этого, была проанализирована встречаемость альтернативных (не ATG) старт-кодонов. Результатом работы стало нахождение белок-кодирующих последовательностей, предположительно подвергшихся положительному отбору и отвечающих за уникальные признаки бактерии.
Введение
Объектом настоящего исследования является бактерия, относящаяся к виду Achromobacter xylosoxidans Yabuuchi and Yano, 1981 рода Achromobacter семейства Alcaligenaceae порядка Burkholderiales класса Betaproteobacteria филума Pseudomonadota [1]. Предположительно, Achromobacter xylosoxidans входит в состав микробиоты ушной полости и желудочно-кишечного тракта человека. Эта бактерия является клинически значимой и может вызывать вспышки внутрибольничных инфекций. Наиболее часто встречается бактериемия у пациентов с внутрисосудистыми катетерами, но также были зафиксированы случаи сепсиса желчевыводящих путей, менингита, пневмонии, перитонита, инфекции мочевыводящих путей, конъюктивита, остеомиелита, некротического панкреатита, инфекции протеза коленного сустава и эндокардита протезного клапана. A. xylosoxidans все чаще обнаруживается в выделениях дыхательных путей пациентов с муковисцидозом и ухудшает течение болезни, что, возможно, связано с воспалительным ответом на липополисахарид и цитотоксический фактор бактерии [2]. Исследование генома генома A. xylosoxidans может увеличить наши знания об этой патогенной бактерии и выявить потенциальные мишени для борьбы с заболеваниями, ею вызываемыми.
Материалы и методы
Для анализа распределения длин белков использовались данные в fasta-формате о белок-кодирующих последовательностях (CDS) геномной сборки ASM1672882v1 из базы данных National Center for Biotechnology Information (NCBI). С помощью методов электронных таблиц Google Sheets были рассчитаны длины всех соответствующих CDS белков, а также медианное значение (см. таблицу S1 Сопроводительных материалов) и была построена диаграмма плотности распределения (см. таблицу S2 Сопроводительных материалов).
Информация о расположении CDS на хромосоме Achromobacter xylosoxidans была выбрана из таблицы feature_table той же геномной сборки в базе данных NCBI. В электронной таблице Google Sheets CDS были отсортированы по координате старт-кодона отдельно для плюс- и минус-цепи, что позволило установить наличие перекрывания и рассчитать его длину для каждой пары последовательно расположенных CDS (см. таблицы S3 и S4 Сопроводительных материалов). Далее эти данные были объединены для построения гистограммы (см. таблицу S5 там же).
В третьей части исследования снова была использована таблица CDS для бактерии Achromobacter xylosoxidans банка NCBI. Она была обработана кодом на языке Python (см. документ Google Colaboratory S6 Сопроводительных материалов), результатом работы которого стала таблица, содержащая информацию о CDS с альтернативными старт-кодонами. Работа с таблицей осуществлялась с помощью инструментов Google Sheets.
Результаты и обсуждение
Длины белков Achromobacter xylosoxidans
Длины белков Achromobacter xylosoxidans
По гистограмме на Рисунке 1 видно, что бóльшая часть белков Achromobacter xylosoxidans имеет длину от 60 до 520 аминокислотных остатков (ак). Внутри этой области наблюдаются два пика: более узкий с максимумом при длине 140–160 ак и более широкий с максимумом при длине 300–340 ак. Также важно отметить, что распределение содержит длинный “хвост”, состоящий из белков длиной до 4887 аминокислотных остатков. Медиана составила 290 ак, что несколько выше среднего значения для бактерий, составляющего около 270 ак [3].
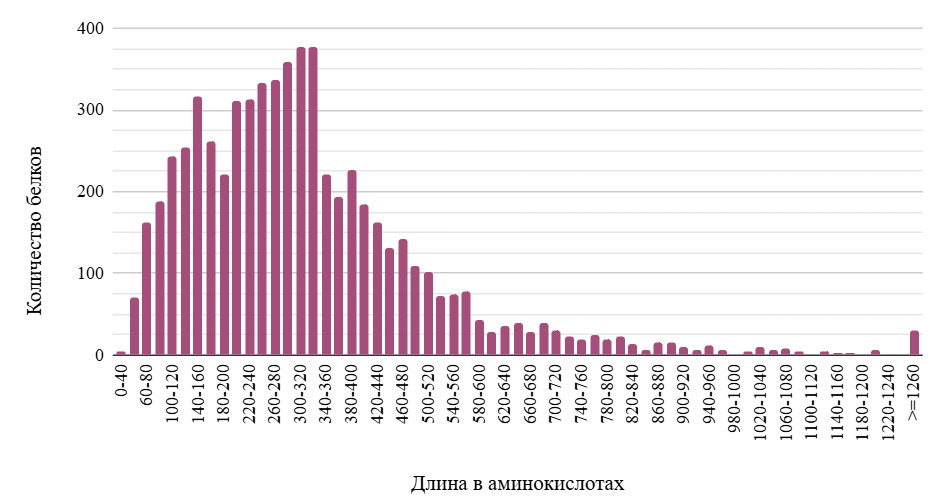
Распределение длин белков у разных организмов носит более консервативный характер, чем другие характеристики генома, такие как количество и GC-состав CDS и распределение изоэлектрических точек белков, и описывается как гамма- или логнормальное распределение с наибольшей плотностью от 50 до 500 ак [3]. Полученное в настоящем исследовании распределение имеет схожую форму. Средняя длина белкового домена равна примерно 100 ак [3], из чего можно сделать вывод, что большинство белков A. xylosoxidans состоят из 1–5 доменов.
Можно предположить, что локальные пики соответствуют многократно дуплицированным генам [3]. Изучение этих генов и белков, в них закодированных, может иметь важное значение для понимания организации и образа жизни бактерии и, следовательно, представляется возможным направлением дальнейших исследований.
Наиболее вариабельной частью распределения длин белков является его правая часть, образующая длинный “хвост”. Белки, имеющие бóльшую длину и более сложную структуру, могут выполнять более специализированные функции и тем самым давать организму некоторое эволюционное преимущество. Однако, чем длиннее белок, тем выше энергетические затраты на его синтез. Таким образом, можно сказать, что длина белков определяется “компромиссом” между данными двумя факторами отбора. Также важно отметить, что очень длинные белки (более 1000 ак) элиминируются в силу экспоненциального увеличения вероятности образования стоп-кодона с ростом длины последовательности и эффекта накопления вредных мутаций [3]. В связи с этим можно предположить, что аномально длинные белки подверглись действию сильного положительного отбора, отвечают за адаптивные признаки и при дальнейшем изучении могут дать важную информацию об особенностях биологии вида Achromobacter xylosoxidans.
Длина пересечений между кодирующими последовательностями на одной цепи
В ходе данного исследования был проведен анализ распределения длины пересечений между белок-кодирующими последовательностями, расположенными на той же цепи хромосомы A. xylosoxidans. Количество перекрывающихся CDS составило 16,4% от их общего числа в геноме. При этом распределения длин на плюс- и минус-цепях не различаются, а почти 78% перекрытий имеет длину один или четыре нуклеотида (см. Рисунок 2), тогда как перекрытий длиной два или пять нуклеотидов нет. Также было рассчитано суммарное количество перекрывающихся областей длиннее пяти нуклеотидов для каждой рамки считывания. Перекрытий белок-кодирующих областей в одной рамке считывания (в фазе 0) обнаружено не было, а соотношение числа перекрывающихся CDS со сдвигом в рамке считывания на один нуклеотид (в фазе 1) к таковому со сдвигом на два нуклеотида (в фазе 2) составило 3,8 (184 к 48).
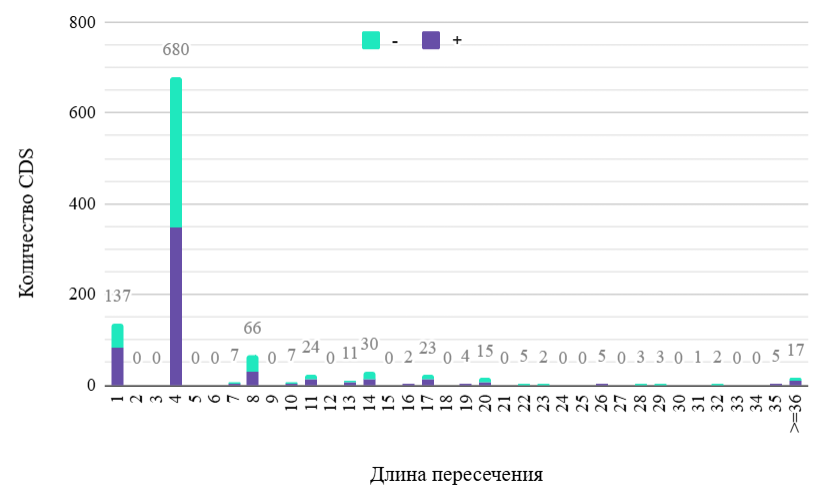
Перекрывания генов обладают эволюционным преимуществом, так как способствуют компактизации генома и позволяют рибосоме непосредственно после терминации трансляции одной открытой рамки считывания в опероне переходить к инициации трансляции другой, что дает бактериям возможность синтезировать функционально связанные белки в требуемом соотношении. При этом перекрывание белок-кодирующих областей не является критичным, но стоп- и старт-кодоны соседних рамок считывания должны находиться на расстоянии нескольких нуклеотидов [4]. Поэтому обратимся к обнаруженным коротким (до 5 нуклеотидов) перекрываниям CDS. Во-первых, можно предположить, что значительная часть этих перекрываний находится внутри оперонов [5], так что изучение этих генов может являться одним из направлений дальнейших исследований генома Achromobacter xylosoxidans.
Во-вторых, такие пересечения имеют место только в фазе 2 сдвига рамки считывания, тогда как CDS в фазе 1 доминируют среди более длинных перекрываний. Число пять было выбрано потому, что в перекрытиях до такой длины включительно стоп-кодон одной белок-кодирующей последовательности пересекается со старт-кодоном другой. В случае исследуемой бактерии такие перекрывания существуют только в фазе 2 (один или четыре нуклеотида). Объяснение отсутствия коротких перекрытий в фазе 1 заключается в том, что последние два нуклеотида стоп-кодонов (TGA, TAG и TAA) не совпадают с первыми двумя нуклеотидами преобладающих старт-кодонов (ATG, GTG и TTG), что резко снижает вероятность перекрытий по два нуклеотида. В пятинуклеотидных перекрытиях первый нуклеотид стоп-кодона вышележащей CDS (во всех случаях T) должен служить последним нуклеотидом старт-кодона нижележащей (G для наиболее представленных старт-кодонов). Таким образом, такие перекрытия возможны только при инициации трансляции нижележащего гена с нетипичного старт-кодона и были показаны для бактерий [6], но для A. xylosoxidans обнаружены не были, что подчеркивает крайне низкую вероятность их возникновения. Для пересечений по одному и четыре нуклеотида таких ограничений нет, что делает их наиболее представленными в геноме бактерии.
Длинные перекрытия, как и длинные белки, должны подвергаться действию отрицательного отбора в силу высокой вероятности образования стоп-кодона или накопления вредных мутаций, и их представленность ожидаемо уменьшается с увеличением длины. Тем не менее, в геноме A. xylosoxidans они были найдены в заметном количестве. Отсюда вытекает следующий вопрос: имеют ли они функциональное значение или всего лишь являются следствием случайных мутационных событий и будут утеряны в ближайшем будущем [5]? Возможно, сравнение генома Achromobacter xylosoxidans c геномами родственных видов прольет свет на эту проблему. Что касается неравномерного распределения числа пересечений по фазам сдвига, оно соответствует гипотезе, предполагающей вклад частоты кодонов. Все старт-кодоны в фазе 2 содержат кодоны TGN (где N — любой нуклеотид) фазы 0, из них TGA — стоп-кодон, который не может находиться в данной позиции вышележащей рамки считывания, а другие три кодируют триптофан и цистеин — одни из самых редких аминокислот. Старт-кодоны в фазе 1 пересекаются с кодонами NAT, NGT и NTT в фазе 0, которые соответствуют средне- или высоко-представленным аминокислотам. Таким образом, значительная доля неравномерности распределения перекрываний по фазам сдвига может быть объяснена структурными причинами, но вопрос о влиянии естественного отбора остается открытым [6].
Встречаемость альтернативных старт-кодонов и длина начинающихся с них последовательностей
Подавляющая часть белок-кодирующих последовательностей у бактерий имеет старт-кодон ATG, которому в процессе биосинтеза белка сопоставляется аминокислота формил-метионин. Однако, было определено, что открытые рамки считывания со старт-кодоном ATG в среднем составляют только 80,1% от общего числа белок-кодирующих последовательностей бактерий. Еще 11,6% начинаются с GTG и 7,8% — с TTG. Было установлено, что эти соотношения очень близки между разными таксонами бактериями [7]. Если сравнивать количество замен в старт-кодонах ATG и метиониновых кодонов внутри CDS, оказывается, что ATG на первой позиции рамки считыванию менее консервативен и, следовательно, меньше подвержен очищающему отбору [8]. Инициация трансляции с альтернативных старт-кодонов происходит по тому же механизму, что и с ATG, что возможно благодаря некомплементарному спариванию оснований, но происходит с меньшей эффективностью.
Было показано, что частота встречаемости неканонических старт-кодонов значительно выше среди коротких рамок считывания (sORFs), которые кодируют белки менее 100 аминокислотных остатков и часто бывают обнаружены в межгенных участках, нетранслируемых областях мРНК, на ‘некодирующих’ РНК. Белки, считываемые с таких последовательностей, в целом изучены плохо, но для них уже были показаны специфические функции. Часто они отвечают за адаптацию к стрессовым условиям, и их экспрессия может индуцироваться изменениями температуры и другими неблагоприятными факторами. Также, если sORF располагается в 5’-нетранслируемой области мРНК, уровень экспрессии белка с нее может влиять на уровень экспрессии основного продукта [9]. Целью данной части настоящей работы был поиск потенциальных коротких рамок считывания в геноме Achromobacter xylosoxidans.
Канонический старт-кодон ATG имели 90,0% проанализированных белок-кодирующих последовательностей. Встречаемость GTG и TTG составила 6,6 и 2,8% соответственно. Также были обнаружены единичные случаи начала рамки считывания с других кодонов (см. Таблицу 1). Представленность альтернативных кодонов оказалась существенно меньше ожидаемой, что может быть связано с недостаточной изученностью и плохой аннотацией генома A. xylosoxidans.
| Старт-кодон | Количество CDS | Процент | q
|---|---|---|
| ATG | 5732 | 90.0 |
| GTG | 422 | 6.6 |
| TTG | 178 | 2.8 |
| CTG | 11 | 0.17 |
| ATC | 9 | 0.14 |
| ATT | 5 | 0.08 |
| ACG | 2 | 0.031 |
| GAA | 2 | 0.031 |
| AAT | 1 | 0.016 |
| ATA | 1 | 0.016 |
| CCG | 1 | 0.016 |
| GAC | 1 | 0.016 |
| GAT | 1 | 0.016 |
| GCA | 1 | 0.016 |
| GCC | 1 | 0.016 |
| GGC | 1 | 0.016 |
| GGG | 1 | 0.016 |
| GTA | 1 | 0.016 |
| GTT | 1 | 0.016 |
| Сумма | 6372 | 100.0 |
| Сумма без ATG | 640 | 10.0 |
В ходе исследования были отобраны CDS с альтернативными старт-кодонами, после чего была определена длина их белковых продуктов (см. таблицу S7 Сопроводительных материалов). Средняя длина последних не отличается от таковой по геному, однако процент коротких (до 100 ак) белков составил 10,0 против 6,7 для всех белок-кодирующих последовательной генома. Также среди них оказалось выше содержание белков, для которых известна только предполагаемая последовательность (15,9% среди CDS с альтернативными старт-кодонами и 11,3% среди всех). Эти данные указывают на возможное присутствие sORFs в геноме A. xylosoxidans в значимом количестве. Дальнейшее изучение этого вопроса может включать исследование локализации CDS c неканоническими старт-кодонами и поиск гомологогичных последовательностей в геномах других видов бактерий.