Сравнение множественных выравниваний
Задание 1
Я написала скрипт для сравнения двух множественных выравниваний. Инструкция вызывается командой python msa.py -h.
Задание 2
Я решила выравнивать домен 10 кДа шаперонина PF00166, с которым я работала в предыдущих практикумах. Я открыла выравнивание seed в Jalview, удалила из него гэпы и выровняла там же тремя программами: Clustal Omega, Mafft и Muscle (проект Jalview). Результаты сравнения трех выравниванием с проверенным людьми выравниванием seed представлены в Таблице 1. Наиболее близким к референсному оказалось выравнивание Clustal Omega c процентом совпадающих колонок немногим более 50. Также можно заметить, что для трех пар выравниваний границы идентичных блоков примерно совпадают, что позволяет нам судить о достоверности выравнивания в этих блоках.
| Выравнивание 1 | Длина выравнивания 1 | Выравнивание 2 | Длина выравнивания 2 | Количество идентичных колонок | Процент идентичных колонок от длины выравнивания 1 | Процент идентичных колонок от длины выравнивания 2 | Координаты идентичных блоков* | Координаты одиночных идентичных колонок** |
|---|---|---|---|---|---|---|---|---|
| Seed | 121 | Clustal Omega | 115 | 62 | 51.2% | 53.9% |
|
15,15 |
| Seed | 121 | Mafft | 120 | 56 | 46.3% | 46.7% |
|
32,32 |
| Seed | 121 | Muscle | 114 | 49 | 40.5% | 43.0% |
|
17,17 |
| *До знака "равно" координаты начала и конца блока в выравнивании 1, после — в выравнивании 2. | ||||||||
| **Координата в выравнивании 1, координата в выравнивании 2. | ||||||||
| Выравнивание 1 | Длина выравнивания 1 | Выравнивание 2 | Длина выравнивания 2 | Количество идентичных колонок | Процент идентичных колонок от длины выравнивания 1 | Процент идентичных колонок от длины выравнивания 2 | Координаты идентичных блоков* | Координаты одиночных идентичных колонок |
|---|---|---|---|---|---|---|---|---|
| Clustal Omega | 115 | Mafft | 120 | 68 | 59.1% | 56.7% |
|
— |
| Clustal Omega | 115 | Muscle | 114 | 54 | 47.0% | 46.4% |
|
— |
| Mafft | 120 | Muscle | 114 | 43 | 35.8% | 37.7% |
|
— |
| *До знака "равно" координаты начала и конца блока в выравнивании 1, после — в выравнивании 2. | ||||||||
Задание 3
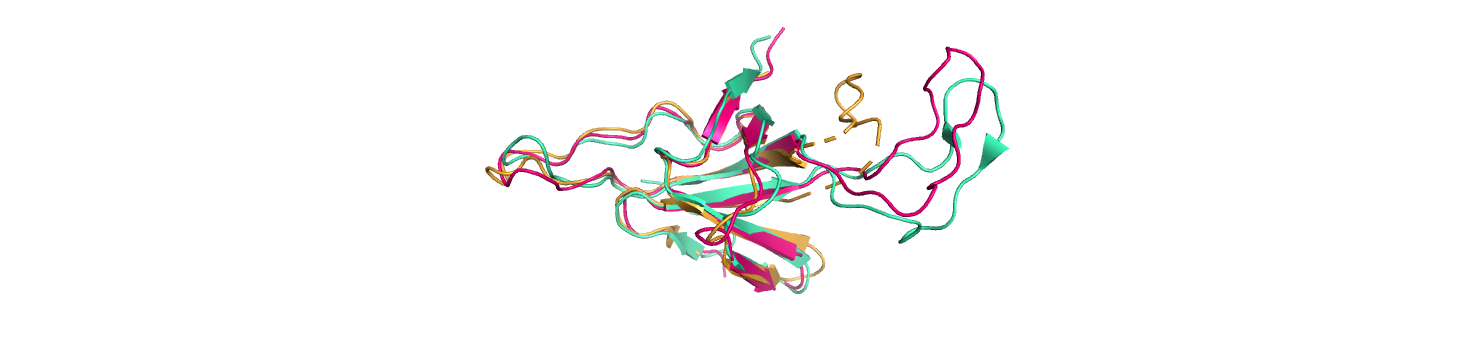
Я выбрала следующие структуры, содержащие исследуемый мной домен: 1AON (цепь O, E. coli), 4PJ1 (цепь O, Homo sapiens) и 1HX5 (цепь A, Mycobacterium tuberculosis). Я создала структурное выравнивание (Рис. 1) в PyMol, взяв структуру 1AON за референсную. Затем экспортировала выравнивание последовательностей, полученное из выравнивания структур, и визуализировала его в Jalview вместе с выравниванием тех же последовательностей программой Clustal Omega (проект). Результаты сравнения этих выравниваний с помощью моей программы отражены в Таблице 3. Выравнивания отличаются примерно на столько же, на сколько различаются множественные выравнивания последовательностей, полученные с помощью разных программ. При этом видно, что выравнивание структур имеет значительно большую длину и, следовательно, подразумевает больше событий образования инделей.
| Выравнивание 1 | Длина выравнивания 1 | Выравнивание 2 | Длина выравнивания 2 | Количество идентичных колонок | Процент идентичных колонок от длины выравнивания 1 | Процент идентичных колонок от длины выравнивания 2 | Координаты идентичных блоков* | Координаты одиночных идентичных колонок** |
|---|---|---|---|---|---|---|---|---|
| Structure | 155 | Clustal Omega | 116 | 68 | 43.9% | 58.6% |
|
|
| *До знака "равно" координаты начала и конца блока в выравнивании 1, после — в выравнивании 2. | ||||||||
| **Координата в выравнивании 1, координата в выравнивании 2. | ||||||||
Задание 4
Clustal Omega — опубликованная в 2011 году программа множественного выравнивания белковых последовательностей. Она относится к быстрым, обладает высокой точностью и позволяет создавать выравнивания более 10 тыс. последовательностей. Она осуществляет построение направляющего дерева не в результате попарного выравнивания всех последовательностей (сложность алгоритма N*N, где N — число последовательностей), а на основе подсчета расстояний каждой последовательности до референсных (их количество пропорционально logN, сложность алгоритма N*logN). Затем последовательности выравниваются по профилям Скрытых марковских моделей (HMM). У этой программы есть функции добаления новых последовательностей к выравниванию и использования существующих профилей НММ (например, указываются в записях Pfam) для выравнивания гомологичных последовательностей.