Практикум 4. ЯМР vs РСА
Задание 1. Вводное
В данном задании сравнивались структуры белка MerB, полученные различными методами. MerB - лиаза, участвующая в системах, отвечающих за резистентность бактерии к ртути (Табл. 1).
| PDB код | 1S6L | 3F0O |
| Метод | ЯМР | РСА |
| Число моделей | 20 | - |
| Разрешение | - | 1.76 Å |
Структура, полученная методом РСА состоит из двух идентичных по последовательности цепей A и B, причём у цепи B больше неразрешённых остатков, и она не имеет в составе лиганд. В модели ЯМР представлена только одна цепь. Было решено сравнивать цепь А модели РСА, т.к. в ней больше разрешённых остатков и модель ЯМР.
В глаза бросается первое различие - отсутствие молекул растворителя в ЯМР модели. Это связано с особенностью метода ЯМР, мы не детектируем сигнал растворителя в отличие от РСА, где можем различить молекулы растворителя, которые кристаллизовались. Также в ЯМР модели представлена структура, усредненная по ансамблю молекул, поэтому мы можем загружить из pdb весь ансамбль и смотреть каждую структуру отдельно.
На макроуровне видим, что в структуре РСА удалось разрешить N-конец - видим альфа-спираль, связанную с анионом брома, а методом ЯМР этот участок на сайте PDB указан, как unmodelled, т.е. структура для этой части не приведена, авторы не смогли достоверно определить структуру этой части, что может быть связано с высокой подвижностью N-конца (Рис. 1), а в РСА этот участок удалось разрешить из-за наличия лиганда, который видимо помогал образовывать стабильную вторичную структуру N-конца и хорошо закристаллизовался. Для C-конца много различных вариантов в ЯМР модели, что опять же может быть связано с высокой подвижностью. Довольно хорошо сопоставляются бета-тяжи в отличие от альфа-спиралей. Также отметим, что неструктурированные участки ЯМР структуры имеют больше возможных конформаций. Вообще структуры не очень хорошо выравниваются друг относительно друга.
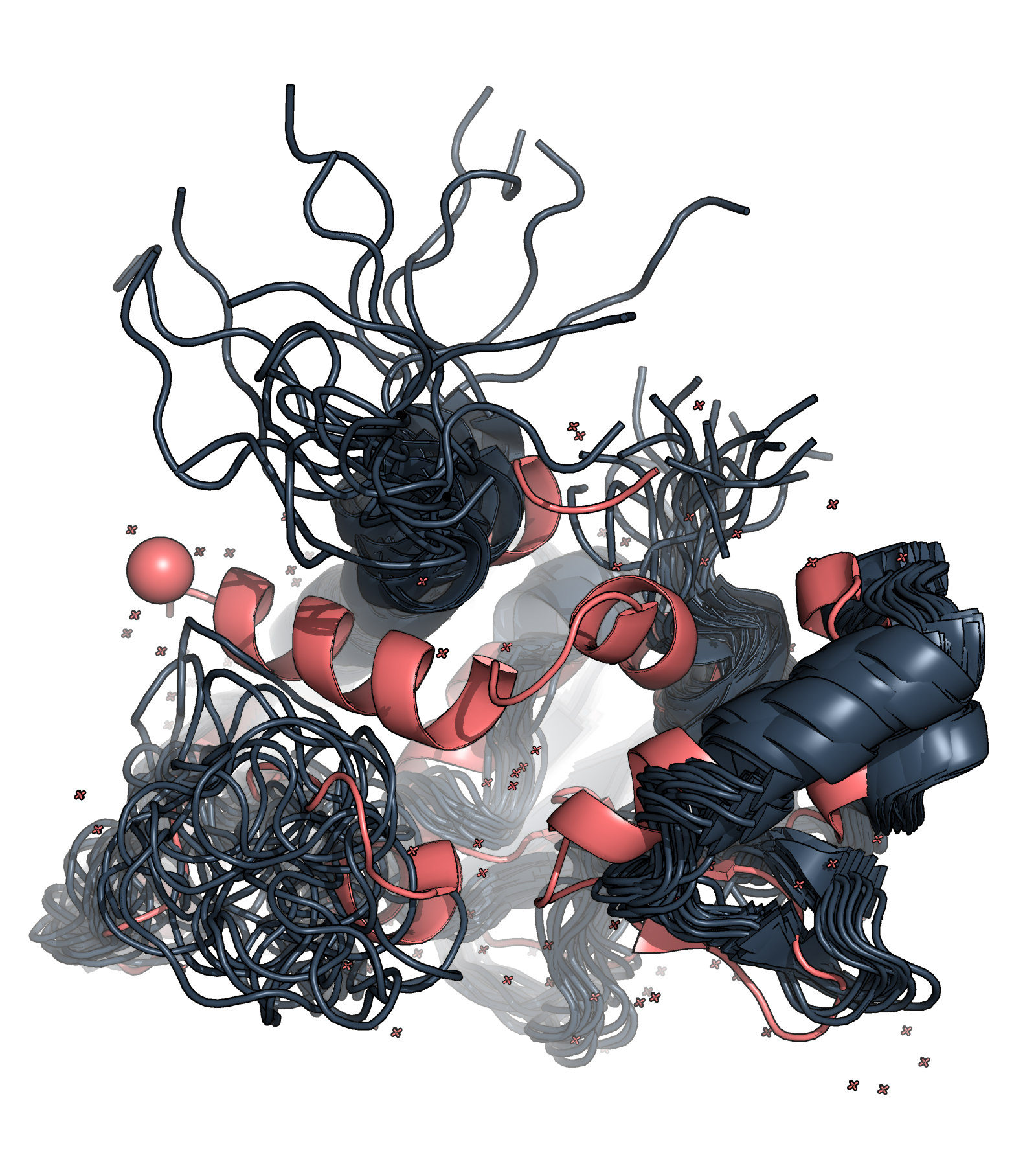
На микроуровне сложно что-то сказать, потому что уже на макроуровне структуры различаются и не очень хорошо выравниваются. Но, например, His-190 в обоих структурах смотрит в разные стороны (Рис. 2). Также на ЯМР структуре можно различить отдельные водороды, чего нельзя сделать в случае с РСА, что связано с особенностью метода ЯМР.
Задание 2. RMSF
Для модели ЯМР можем вычислить меру подвижности отдельных участков - RMSF. В идеальном случае эта мера отражает эволюцию системы во времени - мы получаем набор моделей, которые по мнению авторов больше всего похожи на реальную структуру. Это происходит из-за неполноты данных, которая может быть вызвана шумом в данных или является следствием подвижности. Для структуры, полученной методом РСА, мы можем для каждого остатка вычислить средний B-фактор, который тоже позволяет оценить подвижность белка. Так, мы можем сопоставить эти два параметра и грубо прикинуть, дейсвительно ли ансамбль PDB моделей отражает подвижность белка (Рис. 3).
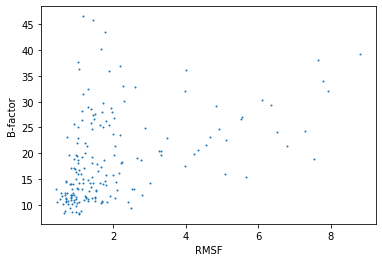
Остатки, которых нет в одной из структур были удалены из рассмотрения. Для каждого остатка вычислены B-фактор и RMSF. В целом наблюдается следующий тренд: чем больше RMSF, тем больше B-фактор, т.е. мы можем судить о подвижности, исходя из усредненных ансамблей модели ЯМР, но делать это нужно с осторожностью, поскольку тут нет однозначной зависимости - имеются остатки с низким B-фактором и большим RMSF и наоборот. Остатки с наибольшим RMSF (около 8) - остатки C-конца белка, который действительно обычно более подвижный, чем глобула.
Задание 3.
В данном задании мы сравнивали водородные связи в структурах, полученных методом РСА и ЯМР. Информация представлена в таблице 1.
| Где водородная связь? | Донор и акцептор | Расстояние в РСА | % моделей ЯМР, где есть эта связь, Å | Min расстояние по всем ЯМР моделям, Å | Med расстояние по всем ЯМР моделям, Å | Max расстояние по всем ЯМР моделям, Å |
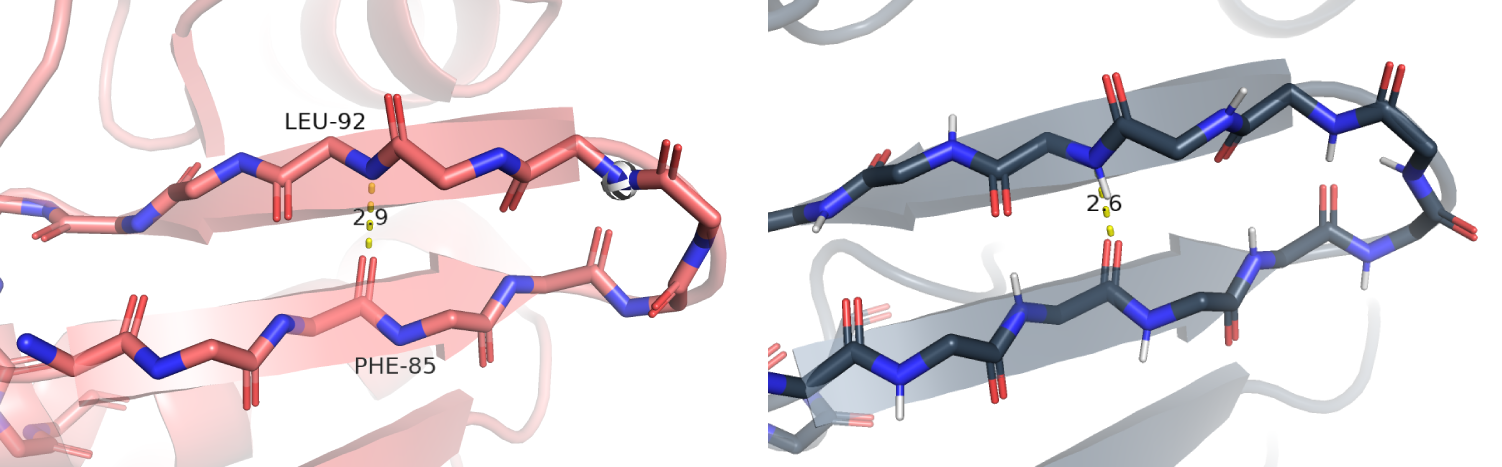
| Между атомами остова в ядре белка | Донор: азот LEU-92. Акцептор: кислород PHE-85 | 2.9 | 100% | 2.5 | 2.5 | 2.6 |
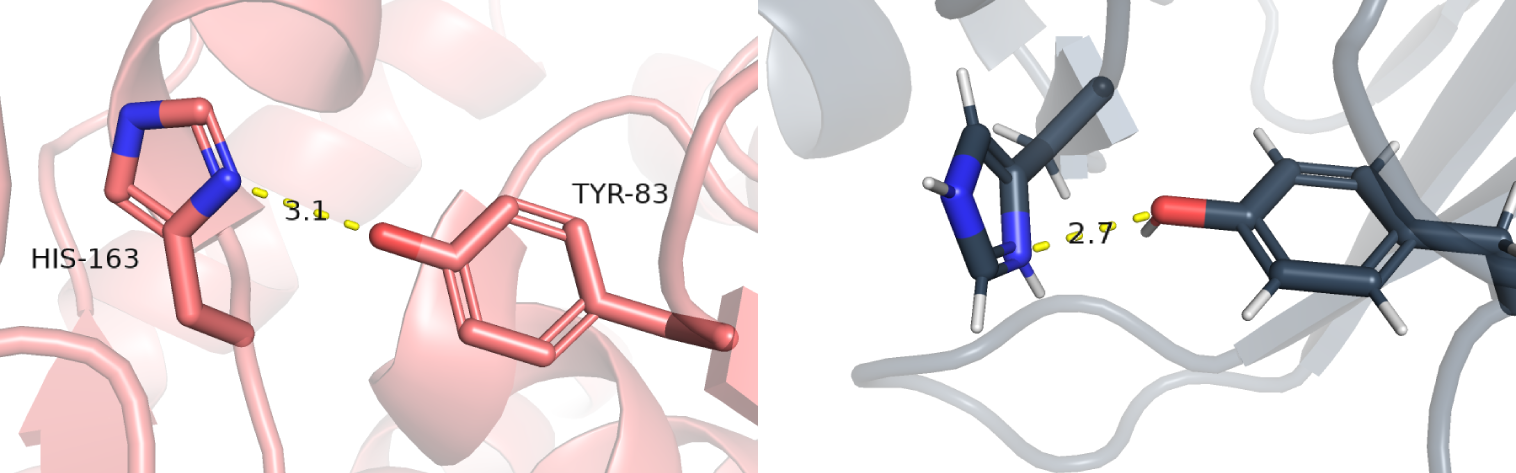
| Связь боковых цепей | Донор: азот HIS-163. Акцептор: кислород TYR-83 | 3.1 | 0% | 2.7 | 4.1 | 5.0 |
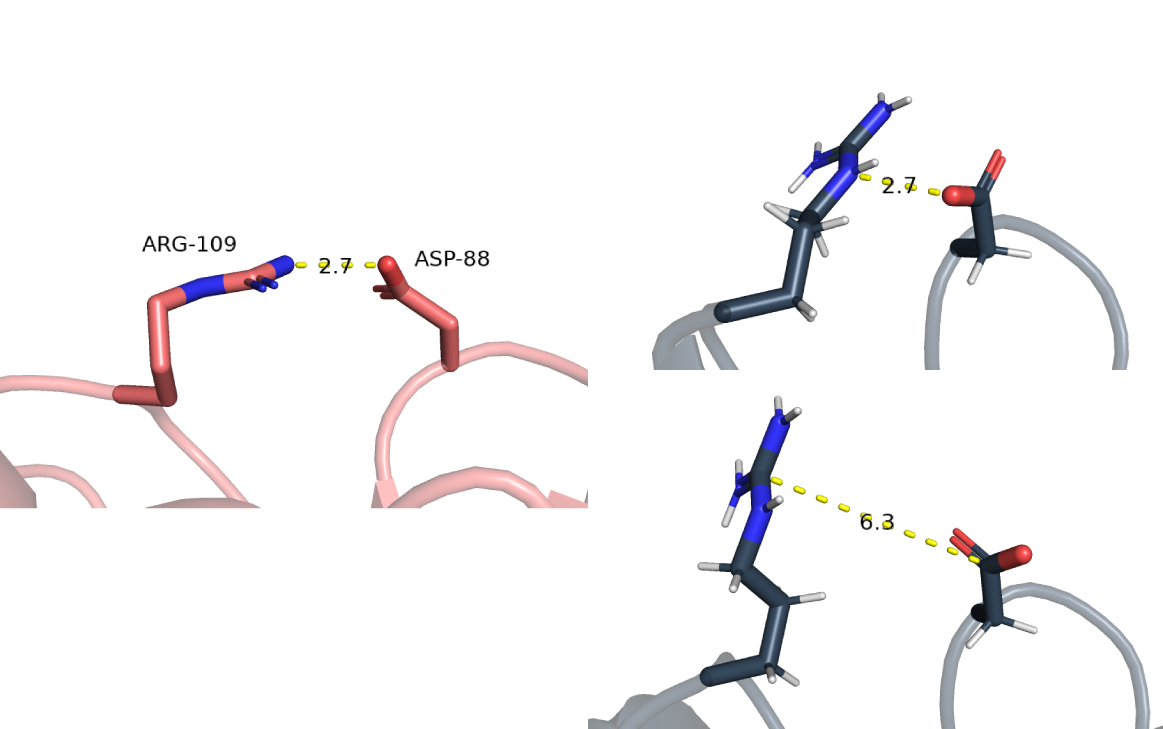
| В петлях, выходящих на поверхность глобулы | Донор: азот ARG-109. Акцептор: кислород ASP-88 | 2.7 | 35% | 2.7 | 3.6 | 6.8 |
Связь между атомами остова в ядре белка
Была выбрана связь между бета-листами. Эта водородная связь сохранилась в во всех ЯМР структурах, что связано с тем, что вторичная структура белка довольно стабильна и остовные водородные связи необходимы для правильного фолдинга белка. Примечательно, что для ЯМР структур длина этой водородной связи была меньше по сравнению с РСА структурой (Рис. 4). Это может быть связано с тем, что в растворе белок ведёт себя по-другому, нежели в кристалле или с особенностью восстановления структуры по данным ЯМР, поскольку у нас уже изначально накладываются ограничения на угол и на расстояние.
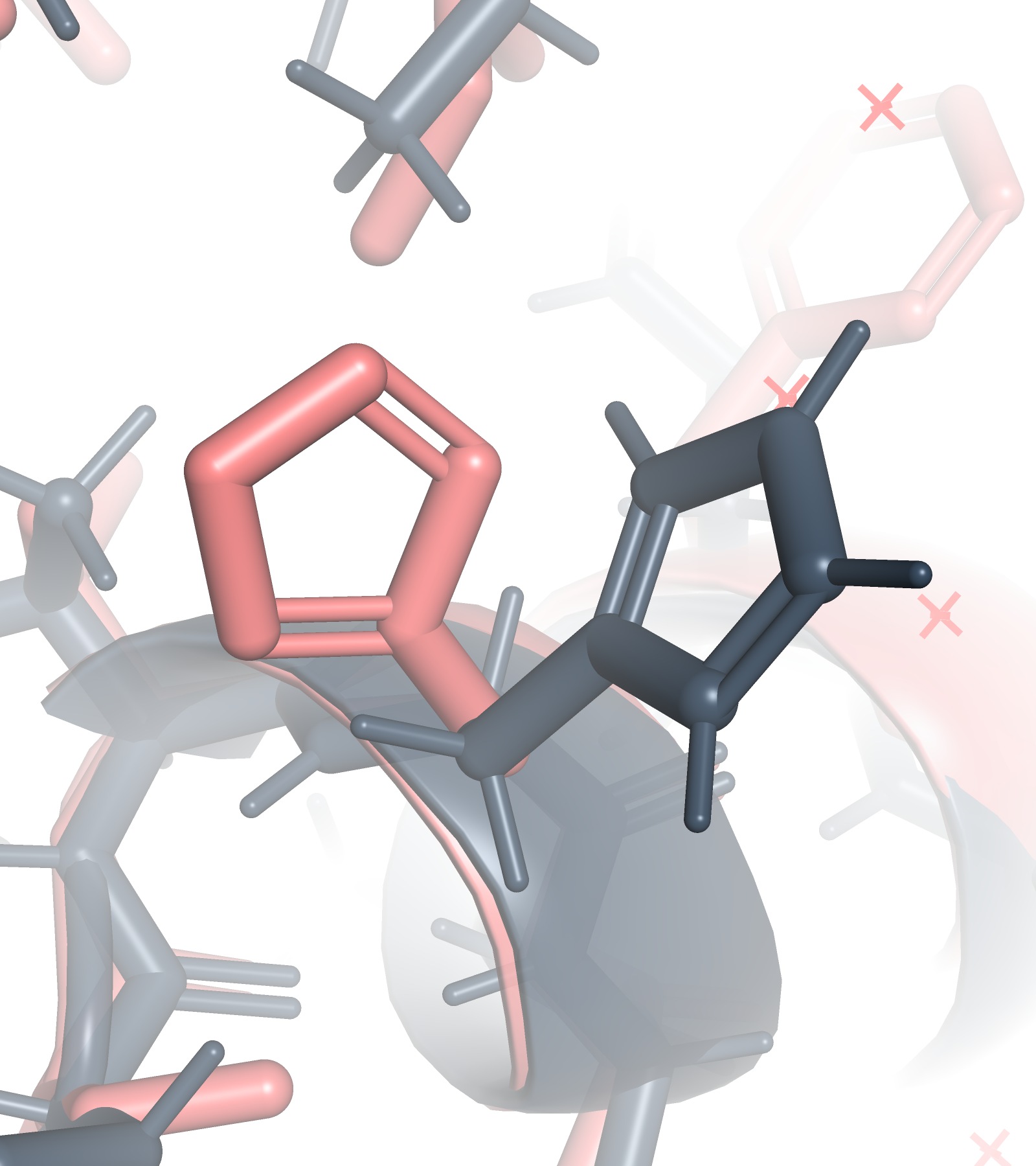
Связь боковых цепей
К сожалению, в ядре белка не нашлось водородных связей между боковыми цепями, т.к. оно состоит преимущественно из гидрофобных остатков, которые не способны образовывать водородные связи. Поэтому были выбраны остатки, которые не являются частью ядра белка. Тирозин, согласно авторской разметке, является частью бета-тяжа, а гистидин - часть петли. В РСА структуре эта водородная связь довольно сомнительна из-за расстояния в 3.1 Å, то в ЯМР структурах мы считаем, что этой связи вовсе нет (Рис. 5). Геометрия молекул такова, что тут скорее всего нет никакой водородной связи, а имеет место какой-то другой вид взаимодействия.
Водородную связь в петлях, выходящих на поверхность глобулы
В случае РСА здесь с высокой вероятностью есть водородная связь, но определить, кто донор, а кто акцептор не представляется возможным, потому что разрешение структуры не позволяет однозначно определить расположение водородов. В случае ЯМР имеет место не самая оптимальная по углу водородная связь. Поскольку аргинин протонирован, то тут скорее всего мы наблюдаем электростатические взаимодействия (Рис. 6). В некоторых случаях эти аминокислотные остатки располагаются довольно далеко друг от друга, что также представлено на рисунке 6. Заметим, что водородная связь сохранилась в 35% случаев, несмотря на то, что это связь в петлях, выходящих на поверхность. Это скорее всего результат того, что Asp-88 является частью бета-поворота, а петля, в состав которой входит Arg-109 соединяет альфа-спираль и бета-лист. Т.к. эти нерегулярные петли расположены между стабильными элементами вторичной структуры, что делает их стабильнее.