Парное выравнивание белков
Парное выравнивание - один из основных методов биоинформатики, основанный на сопоставлении последовательостей двух белков.
Здесь приведено еще одна небольшая работа связанная с аденилат-киназой, в которой используется этот метод.
Мутанты
С помощью скрипта evolve_protein.pl были получены мутантные формы трех участков (по 20 аминокислот) моего белка (аденилат-киназы). Скрипт запускался через Putty следующей командой:
perl evolve_protein.pl –i sw:kad_bacsu –o [файл]
Причем, скрипт запускался трижды с разными параметрами change и replacement. Результаты действия скрипта переводился в формат fasta, после чего добавлялись в jar (в программе Jalview) файл вместе с изначальной последовательностью белка в формате fasta. Затем велся поиск мутированных участков в изначальном белке, и проводилось выравнивание “вручную”.
 |
 |
| Рисунок 1. Выравнивание мутантного участка с показателями change=0.6 replace=0.8. Аминокислоты изначального белка показаны с 26 по 48. |
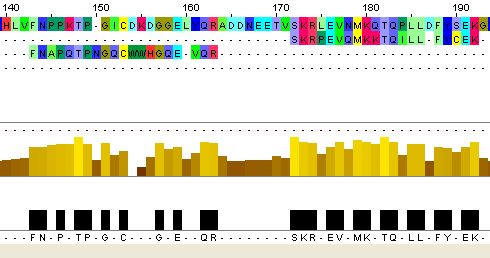
Рисунок 2. Выравнивание участка с показателями change=0.6 replace=0.6 (левее) и участка с показателями change=0.4 replace=0.8 (правее). Аминокислоты изначального белка показаны со 140 по 193. |
| Далее был посчитан процент identity и similarity для каждого выравнивания. Для этого использовалась формула: |
Для similarity принимается m=1 при A(i1)=A(i2) или A(i1)?A(i2). Результаты расчета приведены ниже. |
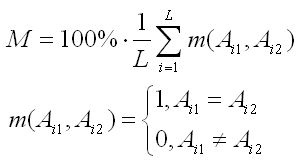 |
 |
Затем был подсчитан все выравниваний с помощью матрицы BLOSUM62.
c=0.6 r=0.8 1+4+4+5-12+6+6+5+6+5+4+0-1-2+5+0-12+5+7-1=45
c=0.6 r=0.8 6+6-1+7+1+5+7-12+6-3+9-4-3-1+6-2+5-12-1+5+5=29
c=0.4 r=0.8 4+5+5-3+5+4+0+5+5+1+5+5-3+4+4-12+6+7-1+5+5=56
Ортологи
Был составлен fasta файл, содержащий последовательность исходного белка (KAD_BACSU) и двух его ортологов (KAD_BACA2 и KAD_BACCQ). Чтобы выровнять последовательности используется функция Web services => Alignment => Muscle with default.
Ны выходе получаем выравнивание тройное выравнивание. Далее из этого выравнивание выделялось 3 парных выравнивания (SU-A2, SU-CQ, A2-CQ). Информация об этих выравниваниях была получена с помощью команды infoalign.
| USA | Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight | Description |
|---|
| fasta::SU_CQ.fasta:KAD_BACSU/1-217 |
KAD_BACSU/1-217 |
217 |
217 |
0 |
0 |
217 |
0 |
0 |
0.000000 |
1.000000 |
Adenylate kinase; |
| fasta::SU_CQ.fasta:KAD_BACCQ/1-216 |
KAD_BACCQ/1-216 |
216 |
216 |
0 |
0 |
156 |
27 |
33 |
27.777779 |
1.000000 |
Adenylate kinase; |
| USA | Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight | Description |
|---|
| fasta::SU_A2.fasta:KAD_BACSU/1-217 |
KAD_BACSU/1-217 |
217 |
217 |
0 |
0 |
217 |
0 |
0 |
0.000000 |
1.000000 |
Adenylate kinase; |
| fasta::SU_A2.fasta:KAD_BACA2/1-217 |
KAD_BACA2/1-217 |
217 |
217 |
0 |
0 |
207 |
4 |
6 |
4.608295 |
1.000000 |
Adenylate kinase; |
| USA | Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight | Description |
|---|
| fasta::A2_CQ.fasta:KAD_BACA2/1-217 |
KAD_BACA2/1-217 |
217 |
217 |
0 |
0 |
217 |
0 |
0 |
0.000000 |
1.000000 |
Adenylate kinase; |
| fasta::A2_CQ.fasta:KAD_BACCQ/1-216 |
KAD_BACCQ/1-216 |
216 |
216 |
0 |
0 |
155 |
27 |
34 |
28.240740 |
1.000000 |
Adenylate kinase; |
Тройственное выравнивание было окрашено по схеме ClustalX, порог identity ntreshold был выбран так, чтобы окрашивались только участки, где минимум две аминокислоты совпадают (он равен 1%).
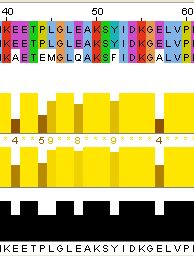 |
Рисунок 3. Участок тройного выравнивания с 40 по 60 аминоксилоту. |
© Марк Меерсон, 2013
Последнее обновление: 22.03.2013