1. Прочтение последовательности ДНК на основании данных, полученных из капиллярного секвенатора по Сэнгеру. Проблемы при чтении хроматограмм.
> Исходные файлы: прямая цепь, обратная цепь.
Файлы были открыты в программе Chromas, и приведены к одному масштабу для удобства сравнения. Для обратной цепи рассматривалась последовательность, комплиментрарная исходной (Edit > Reverse + Complement). Так как первые десятки нуклеотидов хроматограммы обычно не читаемы (например, из-за наличия коротких фрагментов, на которые отжигается праймер[1]), в превую очередь необходимо отделить эти "нечитаемые" участки от основной хроматограммы. После анализа концевых участков хроматограммы прямой последовательности, я пришла к выводу, что с 3' конца "проблемный участок" заканчивается на 24м нуклеотиде (об этом можно судить как по неоднозначности в распределении пиков, так и по количеству неопределенных нуклеотидов, которые не были автоматически распознаны). Далее, несмотря на присутствие шума, последовательность в большинстве случаев определяется однозначно, причем качество хроматограммы сохраняется примерно на одном и том же уровне почти до самого конца(5') последовательности. Вопросы возникают только по поводу последних трёх нуклеотидов, которые я и решила отнести к "нечитаемым". Анализ обратной последовательности показал, что с 3' конца необходимо убрать первые 31 нуклеотида, тогда как с 5' конца нужно было удалить всего 4 нуклеотида. Изображения "проблемных" концов последовательностей представлено ниже.
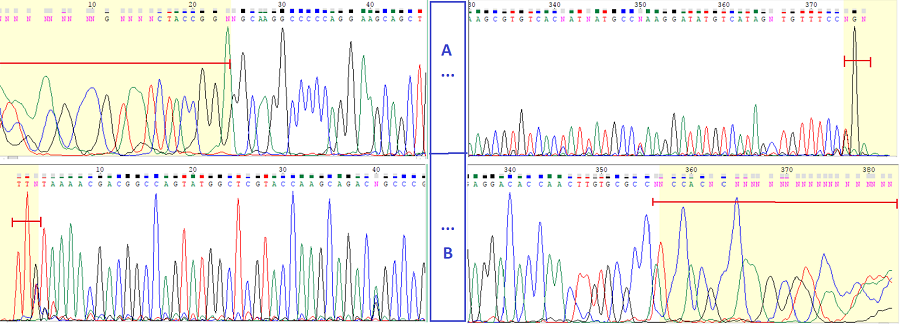
Рис. 1 Проблемные нуклеотиды на концах прямой (а) и обратной (б) поседовательностей. Красным отрезком обозначены границы проблемных областей.
Таким образом становится понятно, что если сравнивать две полученные последовательности друг с другом, можно получить гораздо более точные данные, что я и попытаюсь сделать.
В целом, можно сказать, что качество хроматограмм довольно высоко: большинство пиков определяется однозначно, несмотря на присутсвие шума. Если учитывать редактирование последовательностей относительно друг друга, можно определить все позиции последовательностей. Кроме того, прямая последовательность и последовтаельность, комплиментарная обратной, в основном идентичны. С помощью меода "find" я нашла соответствующие участки на хроматограммах.
Рассмотрим несколько примеров разрешения проблемных нуклеотидов. На "парных" изображениях верхняя последовательность - прямая, нижняя - обратная.
1. На изображении 2 видно, что в прямой последовательности в позициях с 76й по 80ю - определение неоднозначно из-за ошибки хроматографа. Если сравнить эту часть хроматограммы с соответствующим местом в хроматограмме обратной последовательности, можно понять, что нуклеотиды были определены правильно, а значит это место не нуждается в правке.
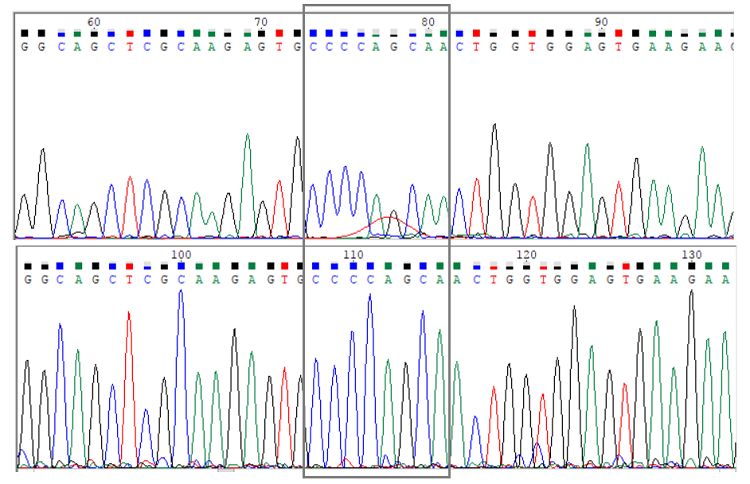
Рис.2
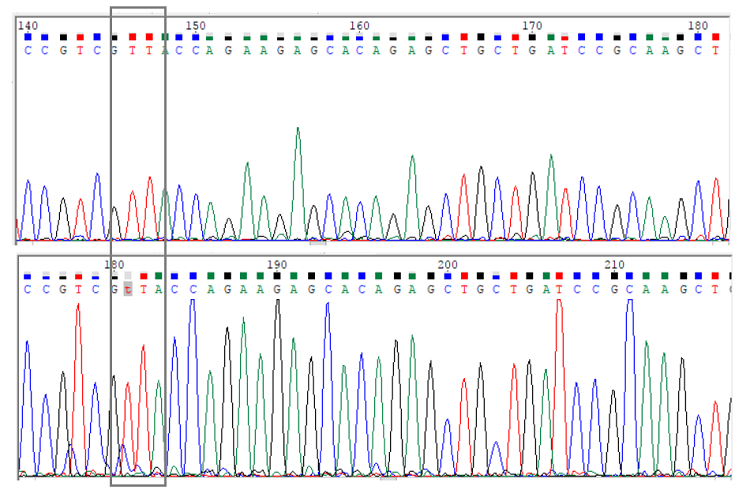
Рис.3
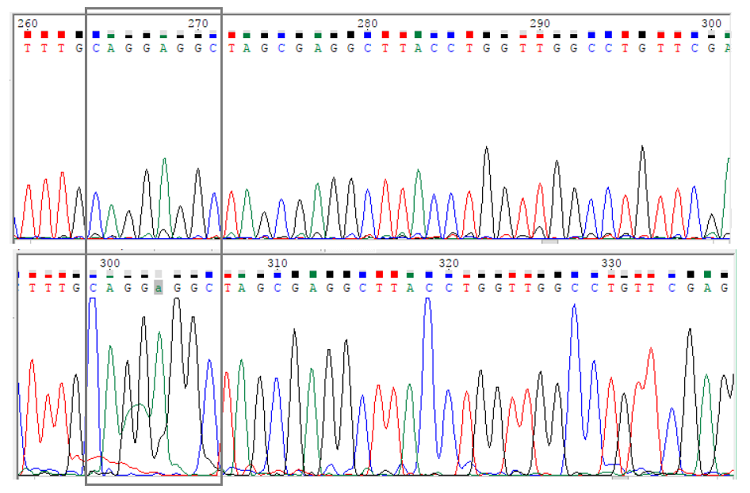
Рис.4
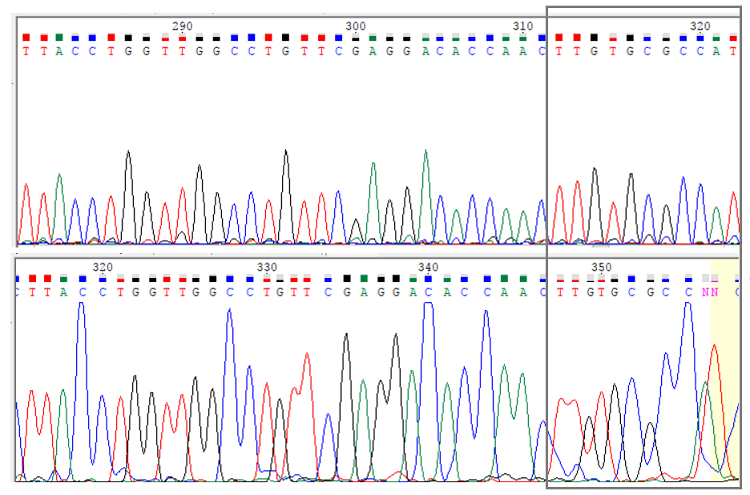
Рис.5
С помощью программы needle, работающей на осове алгоритма Нидлмана-Вунша, я выровняла последовательности по всей длине и импортировала выравнивание в JalView с раскраской по нуклеотидам.
>Ссылка на проект в Jalview
2. Пример нечитаемого фрагмента хроматограммы
Пример плохой хроматограммы я привела ниже.
>Ссылка на скачивание хроматограммы .
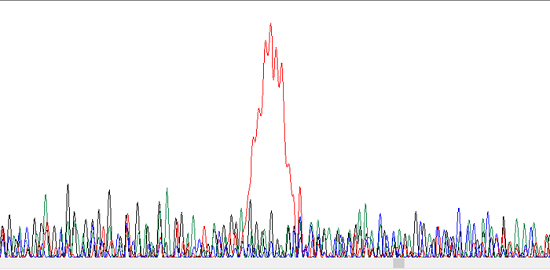
В первую очередь в глаза бросается то, что программа автоматически не определила ни одного нуклеотида. Действительно, величина шума в хроматограмме настолько оставляет желать лучшего, что на хроматограмме нет ни одного однозначно определяемого пика. Я думаю, что такое плохое качество хроматограммы может объясняться в первую очередь чистотой (а точнее не чистотой) образца: возможно, по-ошибке в него попало сразу несколько последовательностей. Помимо этого, в хроматограмме по мему мнению есть что-то похожее на пятно краски хроматографа:
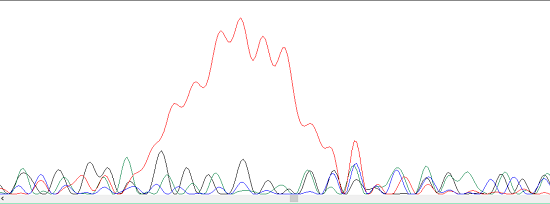
Говорить о каких-либо других ошибках на этой хроматограмме (например, резкое преждевременное падение качества хроматограмы, или неоднозначность предсказания каких-либо нуклеотидов) в данном случае довольно бессмысленно, так как сильный шум нивелирует все возможные эффекты. На этом примере становится ясно, что даже сравнение этого прочтения с обратным не позволит определить последовательность, поэтому делать какие-либо выводы из такой хроматограммы нельзя.