Прочтение последовательностей по Сэнгеру
1. Анализ качества чтений.
Сделан контроль качества чтений с помощью программы FastQC
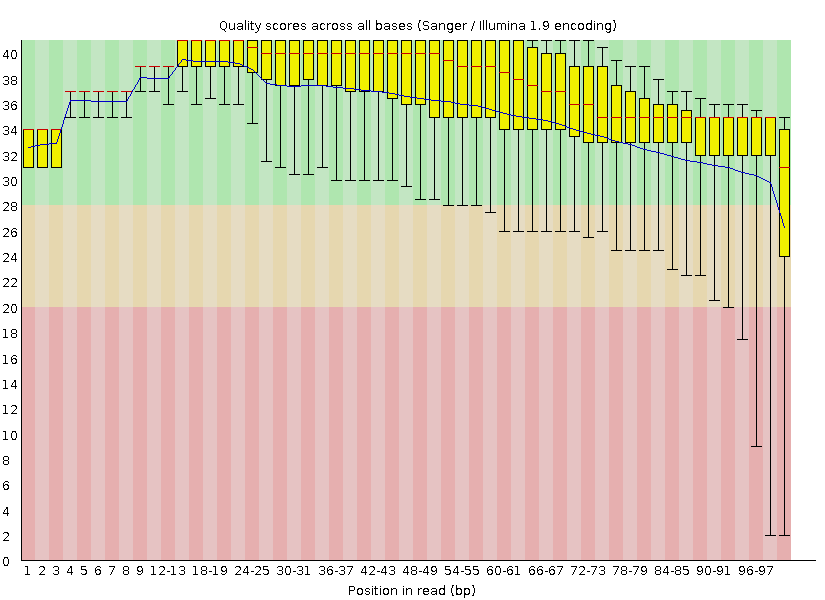
Картинка из FastQC "Per base quality" до чистки Рисунок 1. Per base sequence quality
2. Очистка чтений
Сделана очистка чтений с помощью программы Trimmomatic.
(Отрезаны с конца каждого чтения нуклеотиды с качеством ниже 20, осталены только чтения длиной не меньше 50 нуклеотидов)
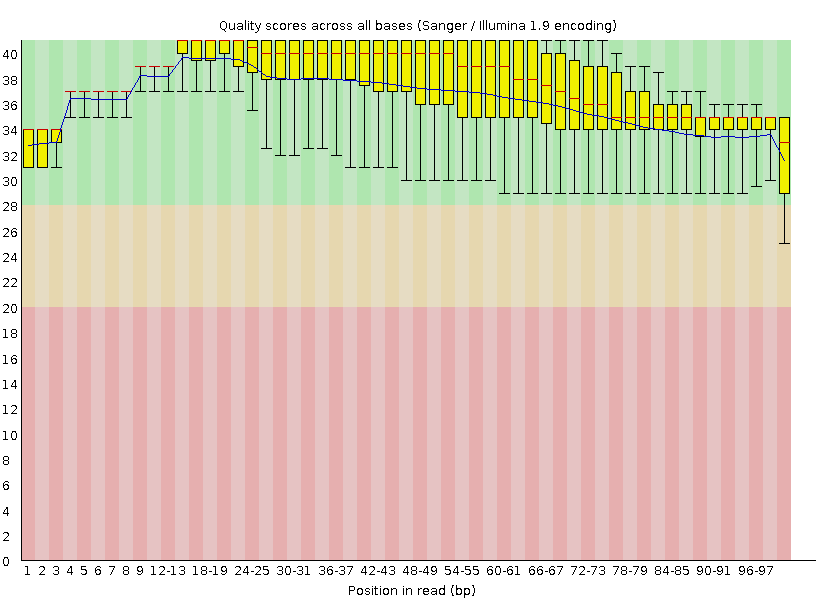
Картинка из FastQC "Per base quality" после чистки Рисунок 2. Per base sequence quality
Таблица команд
| Команда (со всеми параметрами) | Описание (что делает) |
| fastqc chr15.fastq | контроль качества чтений в файле "chr15.fastq" с помощью программы FastQC |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr15.fastq chr15_out1.fastq TRAILING:20 | программа Trimmomatic отрезает с конца каждого чтения в файле "chr12.fastq" нуклеотиды с качеством ниже 20 и сохраняет их в файле "chr15_out1.fastq" |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr15_out1.fastq chr15_out.fastq MINLEN:50 | программа Trimmomatic оставляет из файла "chr15_out1.fastq" только чтения длиной не меньше 50 нуклеотидов и сохраняет их в файле "chr15_out.fastq" |
| fastqc chr15_out.fastq | контроль качества чтений в файле "chr15_out.fastq" с помощью программы FastQC |
Число чтений до чистки: 5068
Число чтений после чистки: 4946
3. Картирование чтений и Анализ выравнивания
Таблица команд
| Команда (со всеми параметрами) | Описание (что делает) |
| hisat2-build chr15.fasta chr15_i | индексирование референсной последовательность в файле "chr15.fasta" с помощью программы Hisat2 |
| hisat2 -x chr15_i -U hr15_out.fastq --no-softclip --no-spliced-alignment -S chr15_ali.sam | построение выравнивание прочтений и референса ("ali.sam") с помощью программы Hisat2 |
| samtools view chr15_ali.sam -b -o chr15_bil.bam | Изменение формата .sam в его бинарный аналог - .bam. с помощью команды samtools view |
| samtools sort chr15_bin.bam chr15_bin.sorted | выравнивание чтений с референсом отсортировано по координате в референсе начала чтения |
| samtools index chr15_bin.sorted.bam | Индексирование отсортированного .bam. файла |
Число чтений, картированных на хромосому: 4935
Число чтений, не картированных на хромосому: 11
4. Поиск SNP и инделей
| Команда (со всеми параметрами) | Описание (что делает) |
| samtools mpileup -uf chr15.fasta chr15_bin.sorted.bam -o poly.bcf | контроль качества чтений в файле "chr15.fastq" с помощью программы FastQC |
| bcftools call -cv poly.bcf -o diff.vcf | программа Trimmomatic отрезает с конца каждого чтения в файле "chr12.fastq" нуклеотиды с качеством ниже 20 и сохраняет их в файле "chr15_out1.fastq" |
три полиморфизма из .vcf файла
| полиморфизм | кордината | тип полиморфизма: замена, вставка или делеция | в референсе | в ридах | глубина покрытия | качество чтений |
| 1 | 89385407 | замена | G | A | 1 | 4.77219 |
| 2 | 89388905 | замена | C | T | 30 | 184.009 |
| 3 | 89382129 | замена | C | A | 11 | 95.0077 |
Всего полиморфизмов найдено - 89. Из них инделей - 2, SNP - 87.
5. Аннотация SNP
Аннотация только полученных snp с помощью программы annovar. Базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
| Команда (со всеми параметрами) | Описание (что делает) |
| convert2annovar.pl -format vcf4 diff.vcf -outfile ch15.avinput | изменение формата файла с полиморфизмами для работы с программой annovar |
| annotate_variation.pl -out refgen_an -build hg19 -dbtype refGene ch15.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация в refgene |
| annotate_variation.pl -filter -out dbnsp_an -build hg19 -dbtype snp138 ch15.avinput /nfs/srv/databases/annovar/humandb.old | аннотация в dbsnp |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000G ch15.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация в 1000 genomes |
| annotate_variation.pl -regionanno -build hg19 -out GWAS_an -dbtype gwasCatalog ch15.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация в GWAS |
| annotate_variation.pl ch15.avinput /nfs/srv/databases/annovar/humandb.old/ -filter -dbtype clinvar_20150629 -buildver hg19 -out CLINVAR_an | аннотация в Clinvar |
При аннотации по refseq мы получили 3 файла refgen_an.exonic_variant_function, refgen.log и refgen_an.variant_function.
В файле refgen.variant_function snp распределены по расположению (локализации): интроны - 72, экзоны - 12, UTR3 - 1, + intergenic(1), + upstrem(3).
SNP попали в гены: LIPC, HMG20A, ACAN
В файле refgen_an.exonic_variant_function содержится информация о синонимичности/несинонимичности snp в экзонах. synonymous - 8, nonsynonymous - 4
У 75 snp есть rs. (DBSnp)
Частоты SNP от 0.00179712 до 0.998602 (1000genoms)
5 snp содержатся в GWAS, т.е. связаны с заболеваниями или предрасположенностью к заболеваниям.
HDL cholesterol 58723426 58723426 A G
Hematological and biochemical traits 58723479 58723479 T C
HDL cholesterol 58723675 58723675 C T
Type 2 diabetes 77777632 77777632 C T
Height 89388905 89388905 C T
Аннотация в ClinVar: 35 het и 54 hom
clinvar выдал пустой файл, значит, информации о связи исследуемых snp с клиническими данными нет
© Grigorjeva Masha