BLAST
1. Определите таксономию и функцию прочтённой вами нуклеотидной последовательности
Для начала с помощью программы consambig была получена консенсусная последовательность из выравнивания прямой и обратной последовательностей.
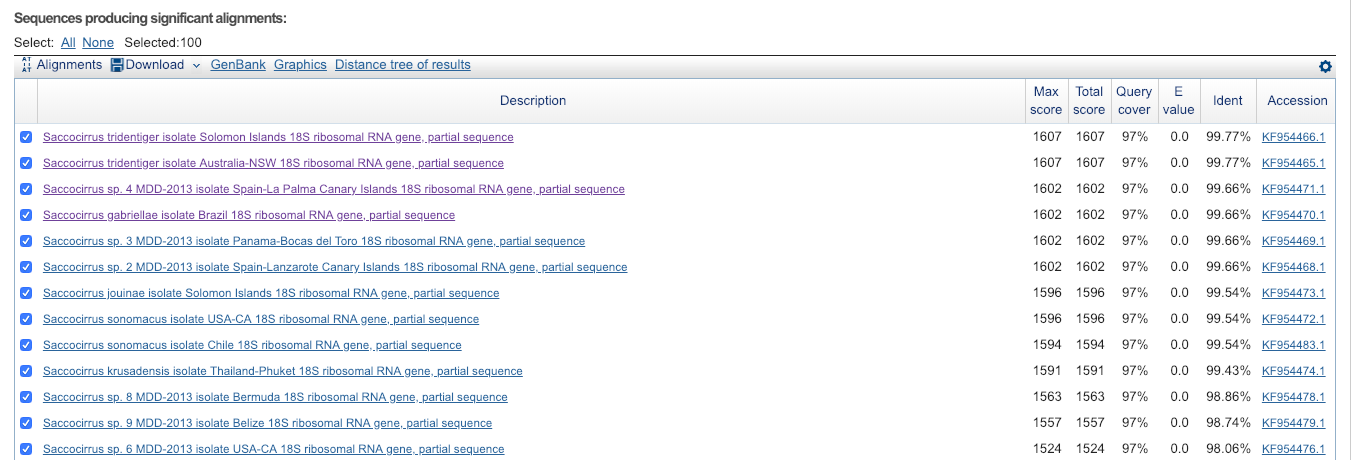
BlastN этой последовательности выдал 100 совпадений. Для определения аннотации взяты 2 лучших совпадения с Ident 99.77% организма Pharyngocirrus tridentiger (Saccocirrus tridentiger). E-value = 0.0. Данные записи соответствуют рибосомальному гену Saccocirrus tridentiger 18S ribosomal RNA gene.
На всякий случай возьмем первое совпедение из предыдущего абзаца и еще одно сосвпадение - с Ident 99.66% организма Saccocirrus gabriellae. E-value = 0.0 Данные записи соответствуют рибосомальному гену Saccocirrus gabriellae 18S ribosomal RNA gene
Ниже приведен скрин выдачи BlastN.
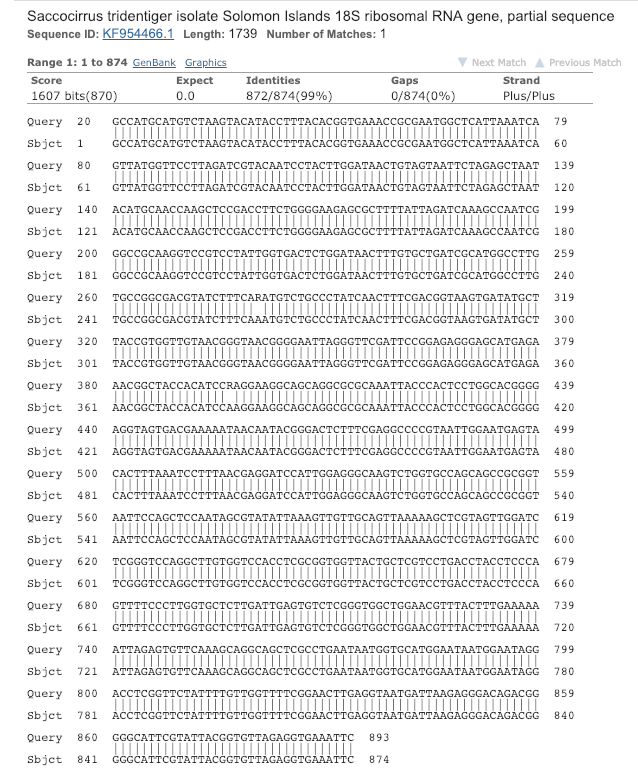
Далее приведено лучшее выравнивание BLAST. Можно заметить, что невыравненных нуклеотидов только 2, оба они вызваны заменами вида A → R или R → A.
Исходя из таких результатов можно с уверенностью сказать, что данная последовательность относится к гену, кодирующему рибосомальную субъединицу 18S рода Saccocirrus, семейства Saccocirridae, класса Polychaeta, типа Annelida
2a. Сравните списки находок нуклеотидных последовательностей pr6 тремя разными вариантами blast
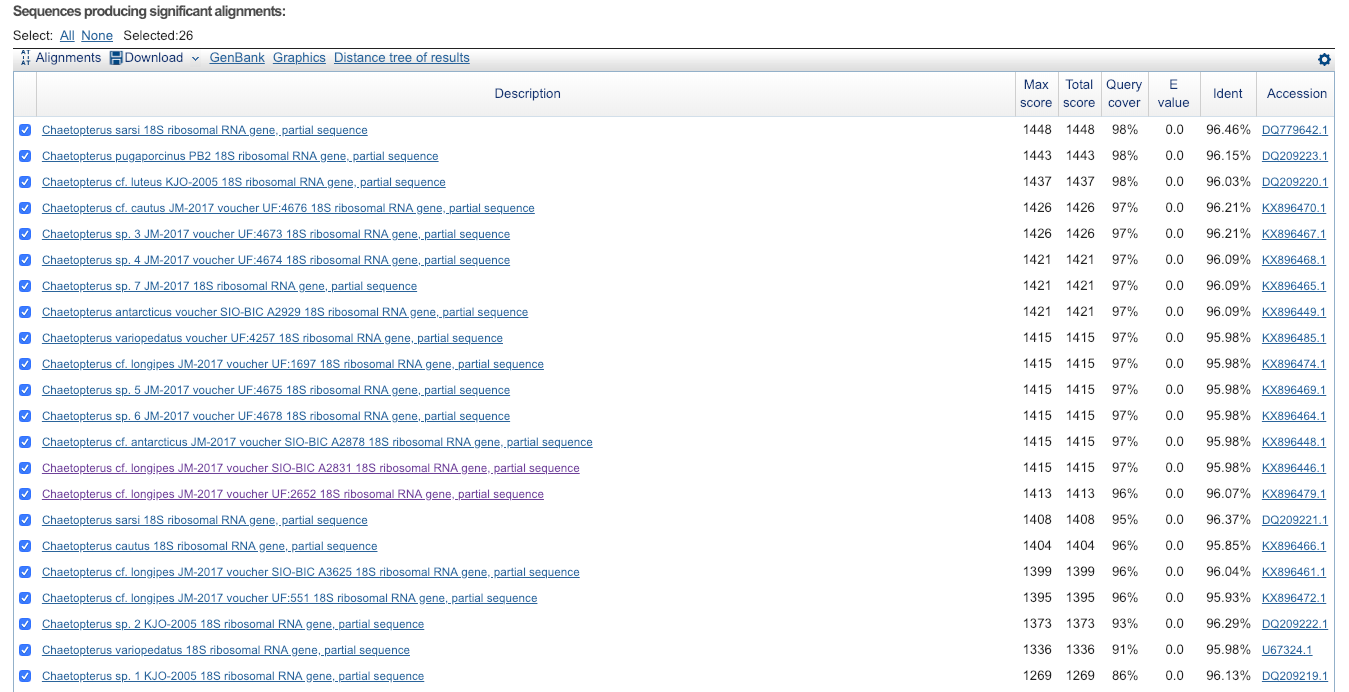
Для выполнения этого задания последовательность из предыдущего пункта не подошла бы, поскольку в условии было указано, что сходство со входной последовательностью должно быть меньше 99%. Для выполнения этого задания я органичивала выборку по роду Chaetopterus
| Алгоритм | Параметры алгоритма | Число находок | Комментарии |
| megablast | Стандартные; длина слова = 28; M/M Score 1,-2; |
26 | Последняя находка с E-value = 2e-172; еще одна (предпоследняя) с E-value = 5e-178, у остальных E-value = = 0.0; поиск ограничен родом Chaetopterus; минимальный Ident = 93.02% |
| blastn default | Стандартные; длина слова = 11; M/M Score 2,-3; |
30 | Последняя находка с E-value = 4.0, таких находок еще 3,помимо этого есть находки с E-value = 1.1, E-value = 8e-174, E-value = 5e-176, у остальных находок E-value = 0.0; поиск ограничен родом Chaetopterus; минимальный Ident = 86.96%; |
| blastn sensitive | Длина слова = 7; M/M Score 1,-4; | 37 | Последняя находка с E-value = 5.9, таких находок еще 9,помимо этого есть находки с E-value = 1.5, E-value = 2e-153, у остальных находок E-value = 0.0; поиск ограничен родом Chaetopterus; минимальный Ident = 93.02%; |
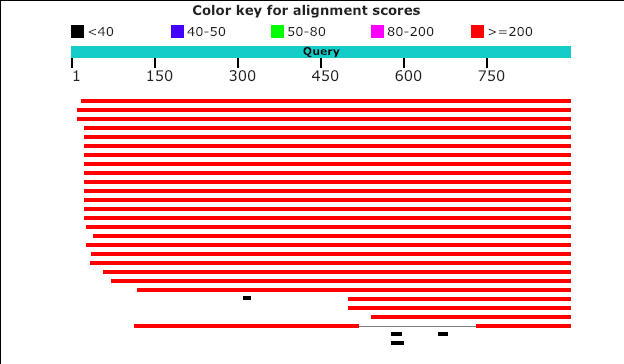
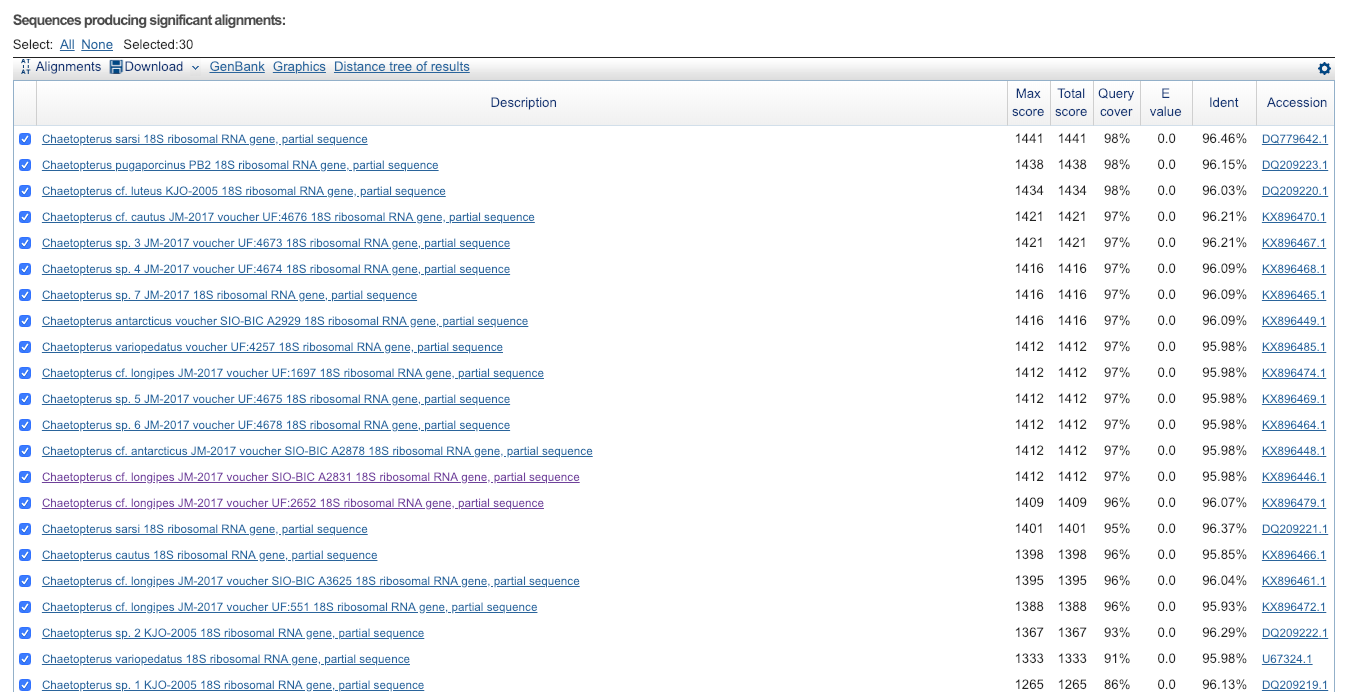
megablast
blastn default
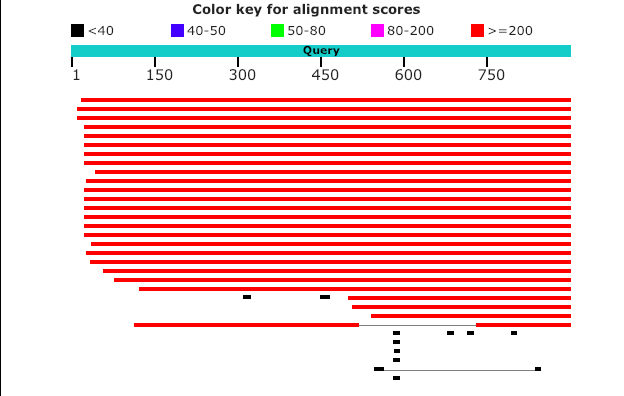
blastn sensitive
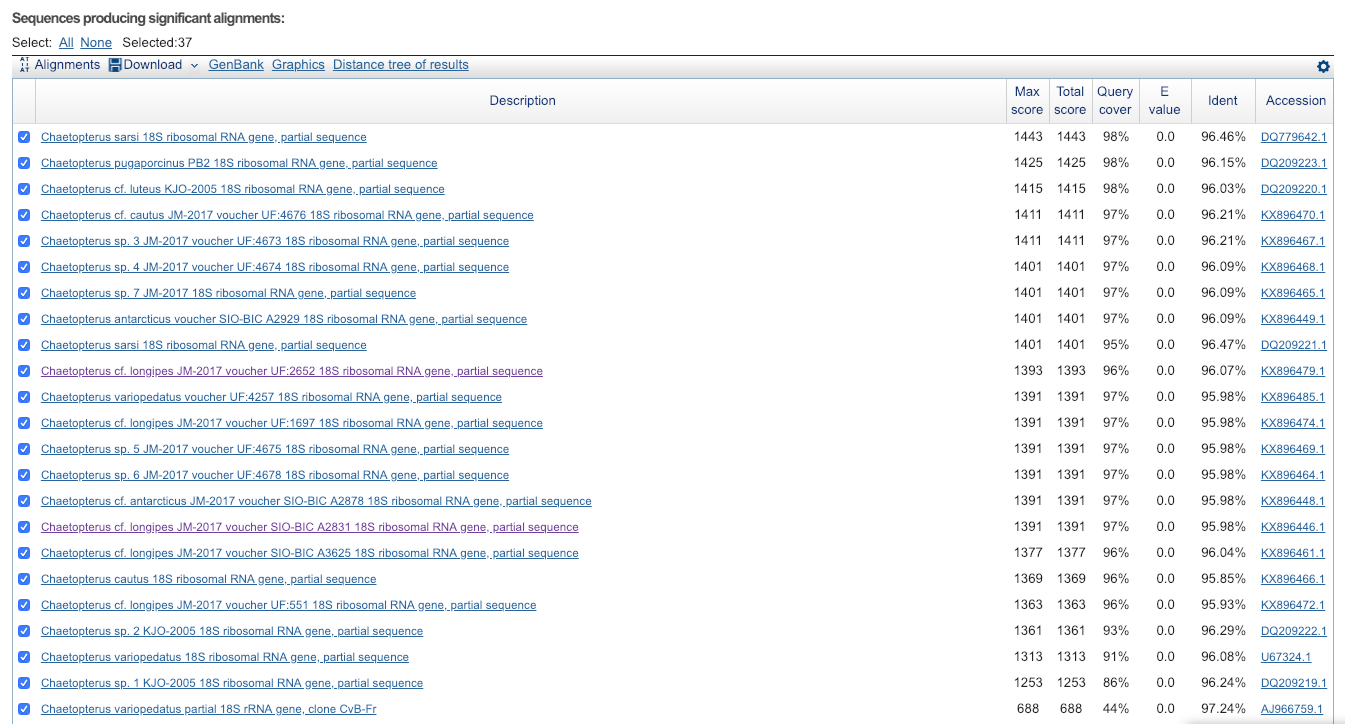
На картинках приведены первые 22 находки (экран не позволял сделать более полный скриншот)
На основании этих данных можно сделать вывод, что blastn sensitive хорошо подходит для точного выравнивания высококонсервативных участков ДНК, а не целых генов, потому что он находит слишком большое количество совпадений с короткой длиной
Megablast стоит использовать при поиске близкородственных организмов с высоким Ident, хотя он пропускает короткие участки из-за word size = 28, с которыми хорошо получается работать у blastn. Количество находок у Megablast наименьшее. Также понятно, что результаты Megablast успешно повторяются другими типами запусков.
Hаиболее низкие значения Е-value наблюдаются при использовании алгоритма ВlastN с параметрами, отличными от стандартных - уменьшение длины слова до 7 в blastn sensitive и изменение значений параметра Match/Mismatch Score до более чувствительных, а вот наибольшие значения Е-value, с небольшой разницей с megablast, показывает BlastN со стандартными параметрами (можно заметить подобную динамику исходя из данных таблицы о количестве находок с E-value больше 0.0, а так же самим значением E-value.
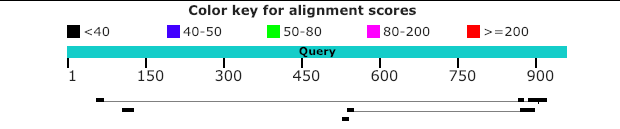
2b . Сравните списки находок нуклеотидных последовательностей pr7 тремя разными вариантами blast
Я провела те же самые запуски только не некодирующей 16S ribosomal RNA 2571..3522 из прошлого практикума (Nippostrongylus brasiliensis)
| Алгоритм | Параметры алгоритма | Число находок | Комментарии |
| megablast | Стандартные; длина слова = 28; M/M Score 1,-2; |
no results | Вывод: "No significant similarity found" |
| blastn default | Стандартные; длина слова = 11; M/M Score 2,-3; |
4 | Последняя находка с E-value = 5.1, остальные находки с E-value = 1.5, E-value = 0.42, E-value = 0.12; минимальный Ident = 82.76%; |
| blastn sensitive | Длина слова = 7; M/M Score 1,-4; | 41 | Последняя находка с E-value = 7.4, таких находок еще 3,помимо этого есть находки с E-value = 1.9 (их пять), E-value = 0.46, E-value = 0.029; минимальный Ident = 94.12%, за ним 94.44%, остальные - 100%; |
blastn default
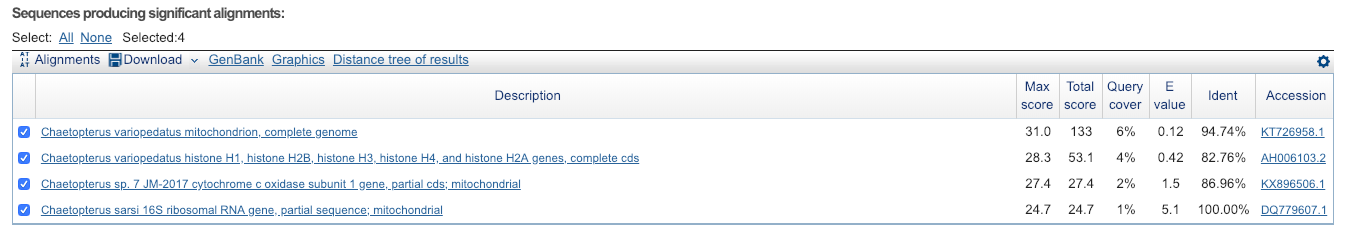
blastn sensitive
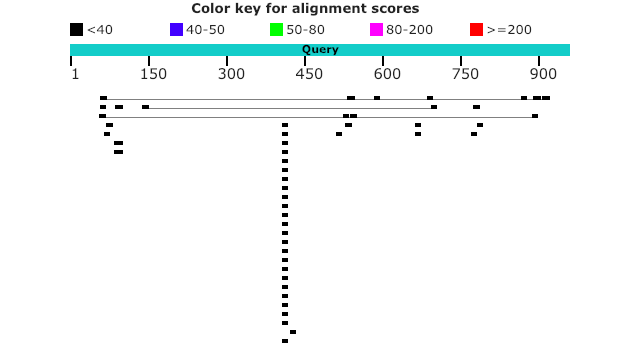
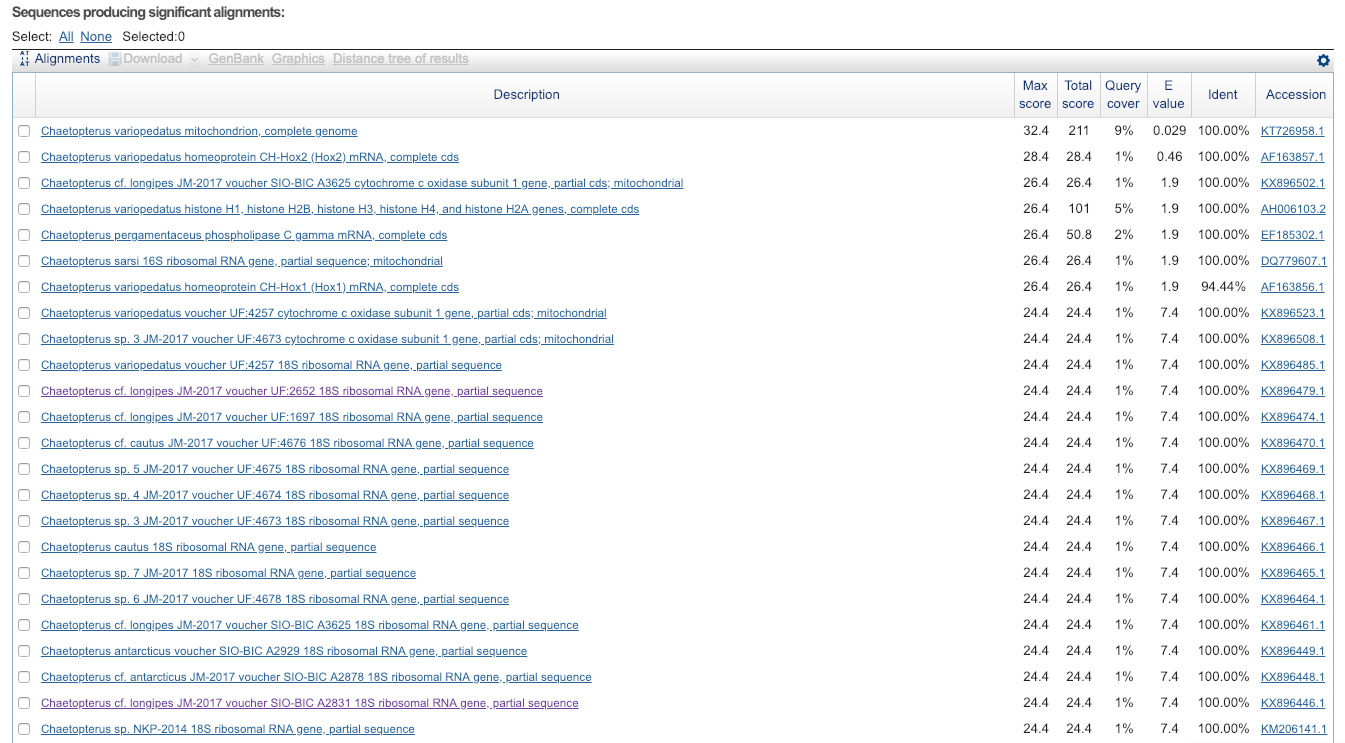
Таким образом мы видим, что MegaBlast может не подойти для поиска гомологии небольших последовательностей, хоть и хорош для быстрого поиска длинной идентичной последовательности.
Этот результат подтверждается так же находками обоих BlastN, тк для всех находок E-value имеет значение больше 0.0. В данном случае находки BlastN default совпадают со значимыми находками BlastN sensitive (как минимум, наиболее вероятная находка, и следующая за ней)
3. Проверьте наличие гомологов трех белков в неаннотированном геноме
Организм Amoeboaphelidium protococcarum (примитивный родственник грибов).
Поиск гомологов проводился при помощи tblastn с порогом на evalue - 0.01
HSP71_YEAST, шаперон HSP70, белок теплового шока
TERT_SCHPO, теломераза,восстанавливающая длину хромосомы при репликации; имеется у большинства (но не всех) эукариот
PRPC_EMENI, митохондриальная цитратсинтаза
Сначала была создана локальная база данных. Команда:
makeblastdb -in X5.fasta -dbtype nucl
Затем с помощью SwissProt я получила последовательности белков в fasta-формате
Далее анализируем белковые последовательности => используем tblastn.
Запрос для HSP71_YEAST:
tblastn -query HSP71_YEAST.fasta -db X5.fasta > HSP71_YEAST.out
Вывод:

Белок: Heat shock protein SSA1 у пекарских дрожжей Saccharomyces cerevisiae (strain ATCC 204508 / S288c). Он выполняет функцию транспорта полипептидов через митохондриальную мембрану и в эндоплазматический ретикулюм, может участвовать в ATP-зависимой разборке в везикулах, покрытых клатрином.
Выравнивание со scaffold-199 имеет E-value = 0.0 и высокий вес (920). Следовательно эти последовательности гомологичны и, похоже, выполняют одну и ту же функцию.
Аналогичный tblastn сделан для TERT_SCHPO и PRPC_EMENI. Ниже приведена сводная таблица для них:
| TERT_SCHPO | PRPC_EMENI | |
| Кодируемый белок | trt1 | mcsA |
| Функция | Теломераза рибонуклеопротеиновый фермент, необходимый для репликации хромосомных концов (удлинении теломер) у большинства эукариот т.е. это обратная транскриптаза, которая добавляет простые повторения последовательности к концам хромосом путем копирования последовательности шаблонов в РНК-компоненте фермента. |
Митохондриальный ген Катализирует синтез (2S, 3S)-2-метилцитрата из пропионил-СоА и оксалоацетата, а также из ацетил-СоА и оксалоацетата с большей эффективностью, имеет активность цитратсинтазы и может заменить недостаток активности citА |
| Лучшее совпадение | scaffold-17 Score: 108 E-value: 1e-23 |
scaffold-693 Score: 393 E-value: 6e-121 |
| Результат | Есть короткие идентичные участки Гэпов нет Чистой гомологии нет, низкий Identity (25%) и Positives(47%) |
Находка условно положительная E-value достаточно низкий, чтобы утверждать гомологию двух белков, но вес недостаточно большой, чтобы однозначно утверждать об одинаковой функции Есть гомологичные участки длинной ~ 25 а.о. |
| TERT_SCHPO |  |
| PRPC_EMENI |  |
4. Найдите какой-нибудь ген белка в одном из контигов
Для поиска выберите один контиг Amoeboaphelidium protococcarum длины порядка десятков тысяч пар нуклеотидов. Интроны у амебоафилидиума короткие, так что ген может поместиться в одном таком контиге.
Поиск проводился с помощью blastx по базе данных "Reference proteins", поиск ограничен таксоном Fungi. В контиге unplaced-10 длиной 37804 пар оснований был найден ген, кодирующий ABC-транспортер (ABC transporter transmembrane region 2-domain-containing protein [Glomus cerebriforme]) с E-value = 0.0
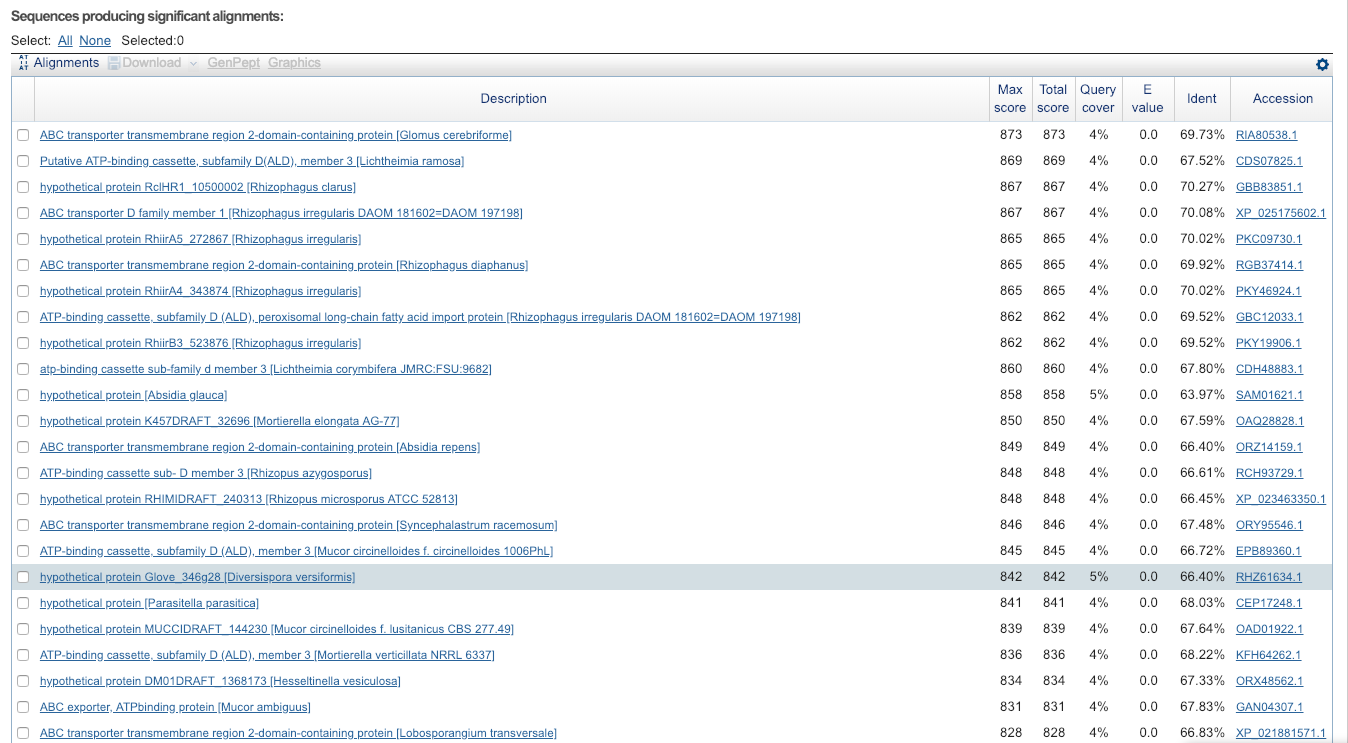
Низкий E-value вызван большой длиной контига и короткой длиной интрона
5. Карта локального сходства геномов двух бактерий
Выберите две бактерии одного рода, для которых доступны полные геномы. Постройте их карту локального сходства с помощью подходящего варианта BLAST. В отчёте приведите изображение карты и описание наиболее заметных перестроек, отличающих один геном от другого.
Возьмем геномы из RefSeq двух бактерий рода Carnobacterium: Carnobacterium sp. 17-4 (NC_015391.1) и Carnobacterium sp. CP1(NZ_CP010796.1)
.
Выравнивание производится с помощью Multiple Sequence Alignment blastn.
Ident = 84.59% E-value = 0.0
Перестройки генома прокариот
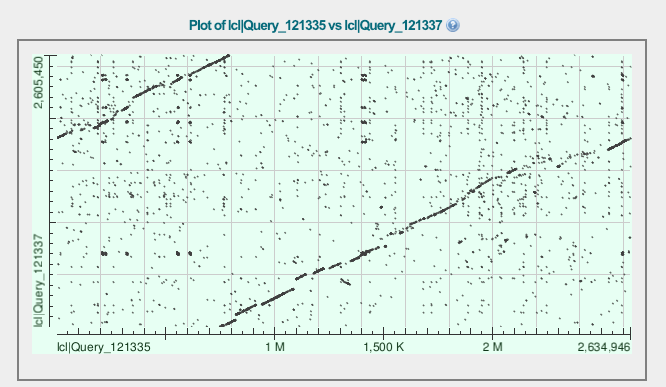
Комментарии: могу предположить, что имеет место смещение точки ориджина, в результате чего получается смещение на графике (левый верхний угол в идеале должен был быть в правом верхнем углу). Подобное расположение куска в левом верхнем углу обсуловлено тем, что что бактерии имеют одну кольцевую молекулу ДНК. Данный участок при развертывании и прочтении просто оказался для одной бактерии в начале генома, а для другой в конце.
© Grigorjeva Masha