Практикум 10
Поиск консервативных мотивов в выравнивании
Для выполнения заданий данного практикума был выбран домен Interferon alpha/beta domain c идентификатором PF00143. Интерфероны I типа (альфа, бета) относятся к более крупному суперсемейству спиральных цитокинов, в которое входят гормоны роста, интерлейкины, несколько колониестимулирующих факторов и ряд других регуляторных молекул.
Анализируемое выравнивание SEED содержало 91 последовательность, что соответствует заданным критериям (более 20, но менее 200 последовательностей). Согласно данным UniProt, этот домен присутствует примерно в 6000 белках, при этом в SwissProt аннотировано только 82 соответствующих белка, что также удовлетворяет условиям задания.
С помощью программы Jalview был осуществлен поиск консервативных мотивов (последовательности на 90% идентичные и более были убраны, однако таких не оказалось). В результате был найден один консервативный мотив с высоким IC (столбцы 163-167). На основе данного сида вероятный паттерн данного мотива может быть следующим в формате Jalview: S[DSRHP]CAW.
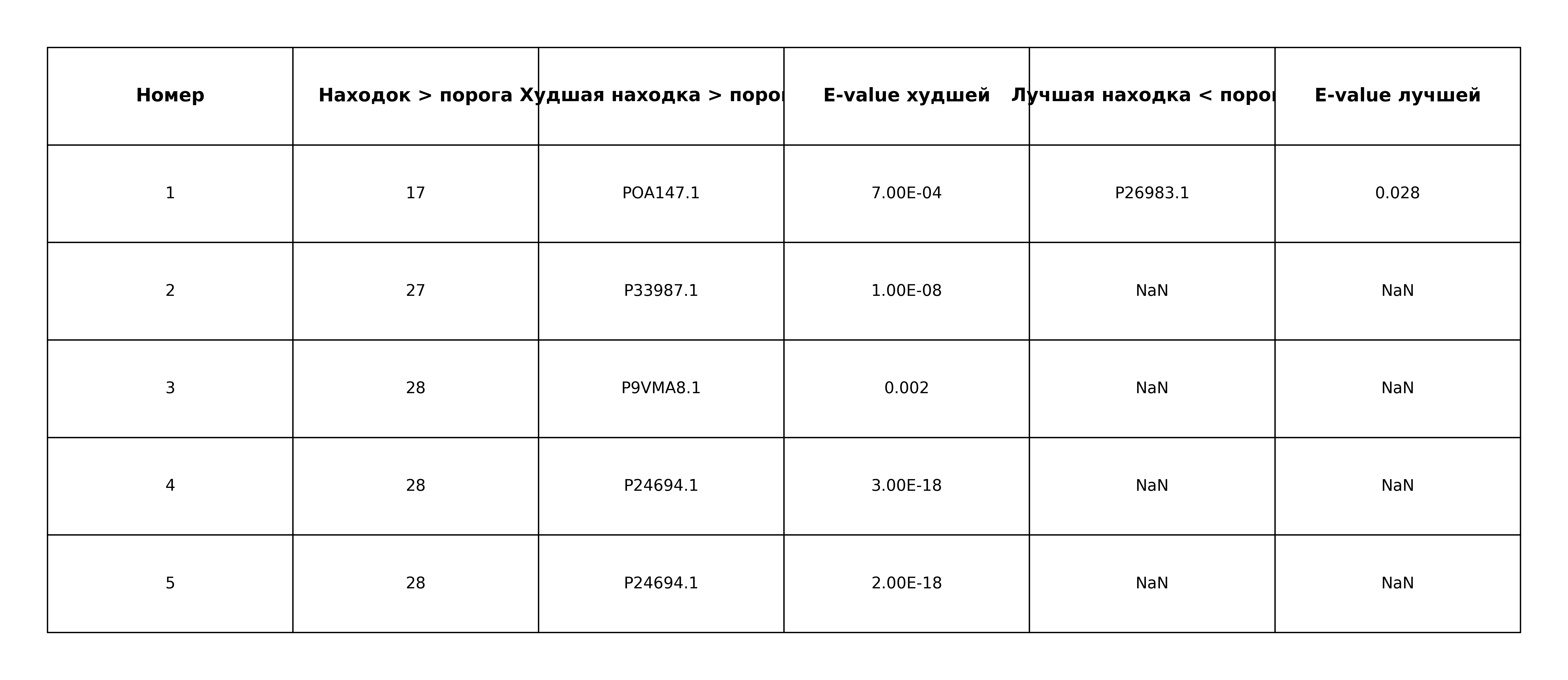
Выполним в Jalview (при помощи find) поиск по паттерну S[DSRHP]CAW во всем выравнивании. Было найдено 63 находки, все друг под другом (всего 91 последовательность).
Переведем паттерн в формат Prosite: S-x-C-A-W. По этому паттерну был проведен поиск на сайте ScanProsite. В результате было найдено 101 находкa в 101 последовательности. Ссылка на результаты поиска.