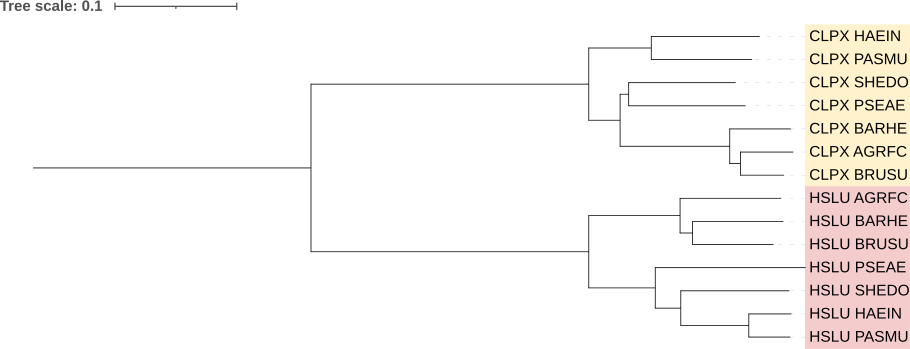
Составление списка гомологичных белков, включающих паралоги
Для выполнения практикума мной были выбраны бактерии:
- AGRFC - Agrobacterium fabrum
- BARHE - Bartonella henselae
- BRUSU - Brucella suis
- HAEIN - Haemophilus influenzae
- PASMU - Pasteurella multocida
- PSEAE - Pseudomonas aeruginosa
- SHEDO - Shewanella denitrificans
Далее в отдельной директории выбранные протеомы были объединены в один файл. Затем на основе выбранных протеомов была создана локальная база данных для запуска blastp при помощи команды:
makeblastdb -in db.fasta -dbtype prot
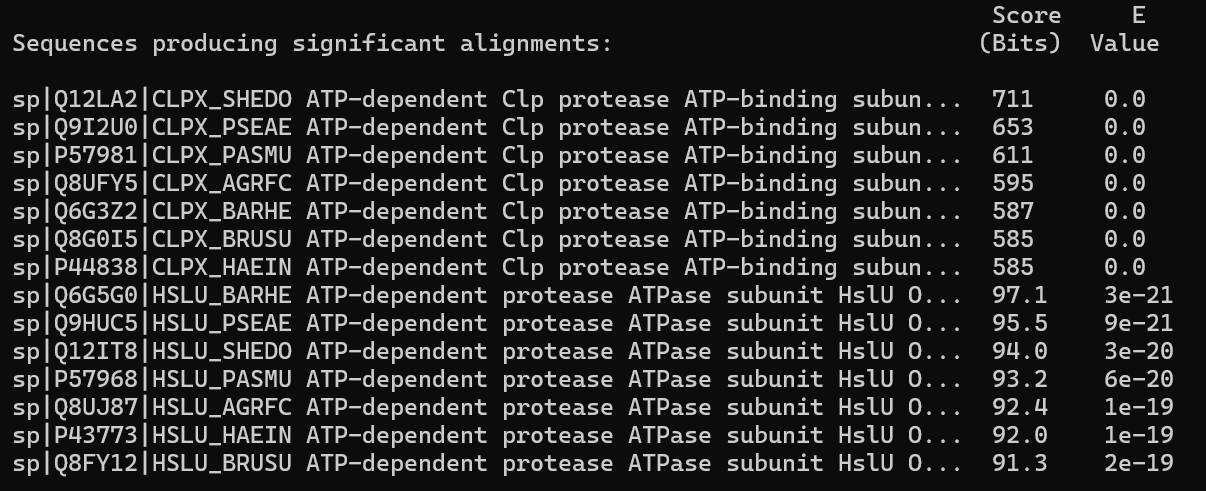
После чего был запущен blastp на основе созданной базы данных. В качестве запроса была подана последовательность белка CLPX_ECOLI (файл). Для отбора достоверных гомологичных белков среди отобранных бактерий был поставлен порог на E-value в 0.0001.
blastp -task blastp -query ref.fasta -db db.fasta -out blast.out -evalue 0.0001
Полная выдача доступна по ссылке. Был получен список из 14 находок: