Введение в транскриптомный анализ
Подготовка референса
В этот раз я все еще работаю с 15 хромосомой, которую теперь проиндексировал при помощи hisat2:
hisat2-build chr15.fna chr15_hi
При данном на вход файле в fasta-формате, на выход были получены 8 бинарных файлов в расширении ht2.
Оценка качества чтений
Как и в одном из прошлых заданий, качество полученных чтений анализируются при помощи fastqc:
fastqc SRR2015718.fastq.gz
Удобный html-формат выдачи можно найти тут.
Судя по полученной странице, общее число чтений составило 33065729.
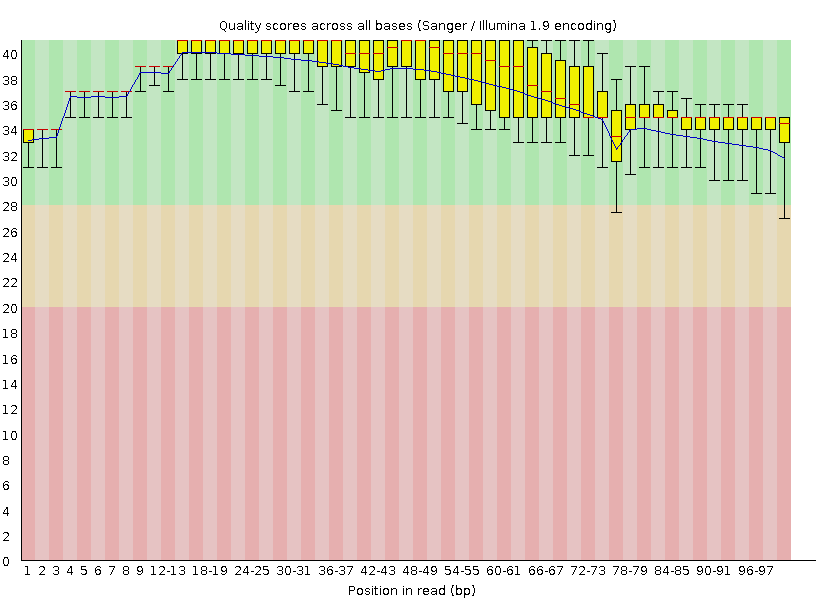
Per base sequence quality
Судя по рис. 1, почти все чтения имеют качество выше 28, что говорит о хорошо проделанной работе по секвенированию. Примечательно, что в среднем качество падает в начале и к концу чтения, что связано с издержками метода.
Sequence Length Distribution
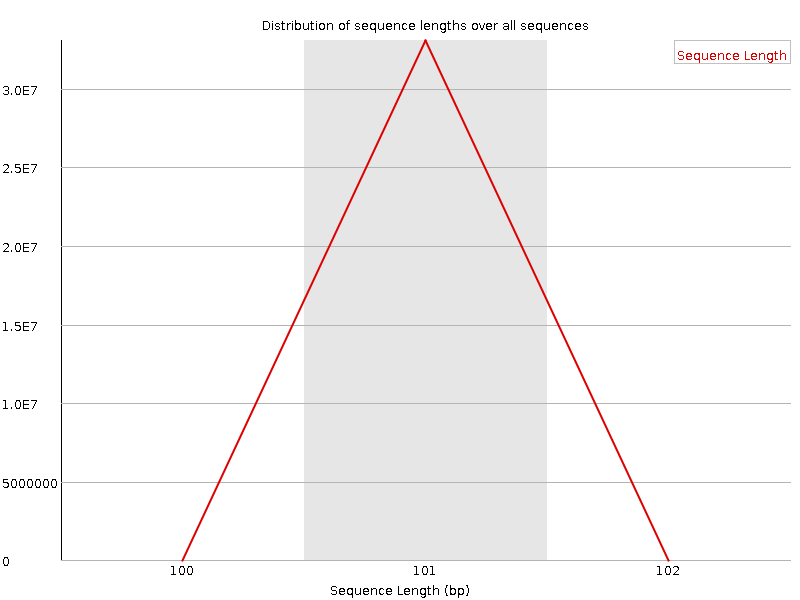
На рис. 2 видно, что абсолютно все чтения имеют длину 101 bp - типичный результат при секвенировании на Illumina.
Картирование чтений на референс
Для картирования снова воспользуемся hisat2:
hisat2 -p 15 -x chr15_hi -k 3 -U SRR2015718.fastq.gz > rna.sam 2> rnalog.txt
Тут "hisat2" - сам алгоритм, "-p 15" - команда на использование 15 ядер, "-x chr15_hi" задает референс, "-k 3" задаёт граничное значения веса выравнивания чтения на референс, "-U SRR2015718.fastq.gz" задает сам картируемый файл. Полученный лог можно просмотреть тут. Судя по нему, все чтения непарные (ну конечно, это и было нам дано), а на референс было картировано хотя бы один раз 1043710 (3,16%) чтений. Похоже, с 15 хромосомы транскрибируется не очень много РНК.
Далее нужно перевести sam файл в сортированный bam и проиндексировать. Для этого использованы следующие команды:
samtools sort -o bam -@ 15 rna.sam -o rna.bam
samtools index -b -@ 15 rna.bam rna.bai
Далее были отобраны только чтения, легшие на 15 хромосому, командами:
samtools faidx chr15.fna -o chr15.fai
samtools view -h rna.bam NC_000015.10 > rna.chr15.sam
samtools view -bS rna.chr15.sam > rna.chr15.bam
Поиск экспрессирующихся генов
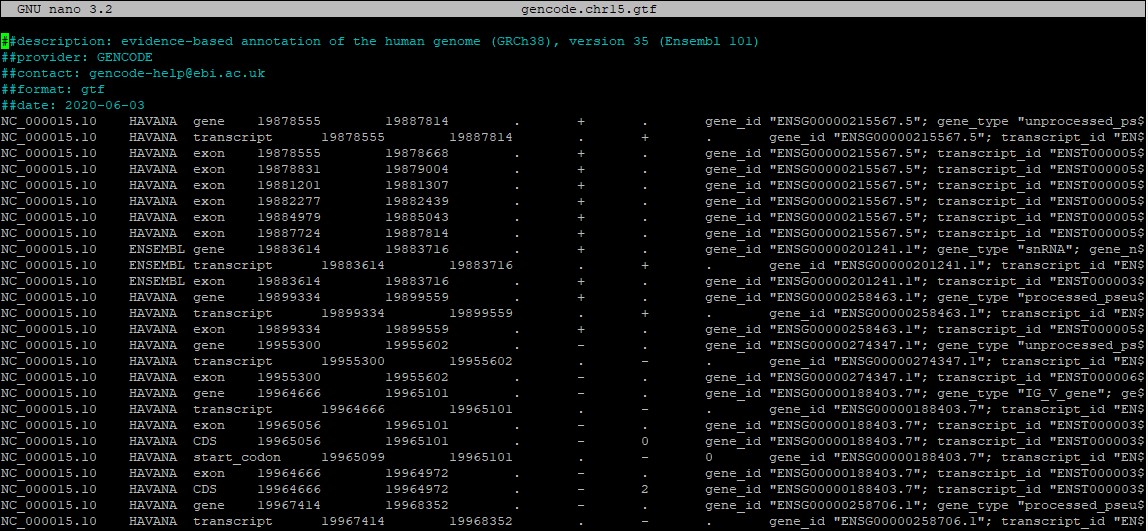
Файл с разметкой генов хромосомы (рис. 3) включает шапку с описанием файла и таблицу генов со следующими столбцами:
*за "+" принято направление 5'-3'
Посчитать число генов на хромосоме помогла команда
awk ' $3 == "gene" ' gencode.chr15.gtf | wc -l
И, если верить выдаче, на 15 хромосоме 2222 гена.
Теперь для каждого гена из разметки нужно посчитать число картированных на этот ген чтений с помощью программы htseq-count:
htseq-count -f bam -s no -m union -t exon rna_reads.bam gencode.chr15.gtf > htseq15.txt
Тут "htseq-count" - непосредственно программа, "-f bam" задает формат вводимых данных, "-s no" заставляет считать считывания перекрывающимися с объектом, "-m union" - режим для обработки чтений, перекрывающих несколько генов, с их объединением.
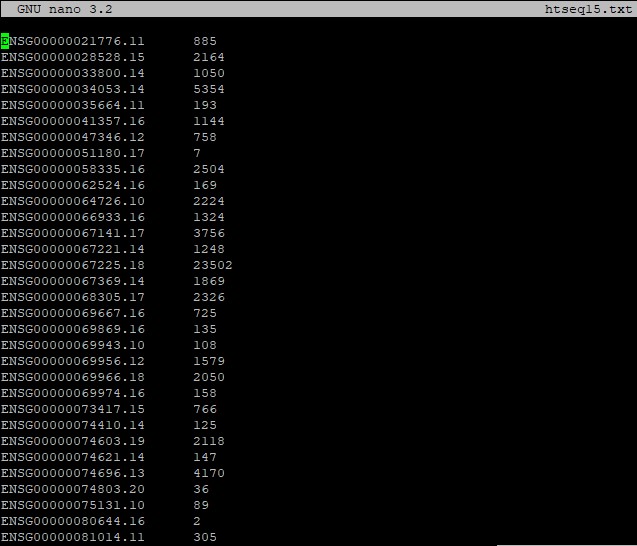
Полученный файл (рис. 4) содержит сопоставление идентификатора гена и числа картирований на него в двух стролбцах.
Для вычисления числа попавших в границы генов чтений, воспользуемся командой:
wc -l htseq15.txt | head -n $lines-5 htseq15.txt | awk '{s+=$2}END{print s}'
Похоже, что таких чтений 729520. А вот информация о попавших мимо генов чтениях находятся в конце файла htseq15.txt, и их 179831.