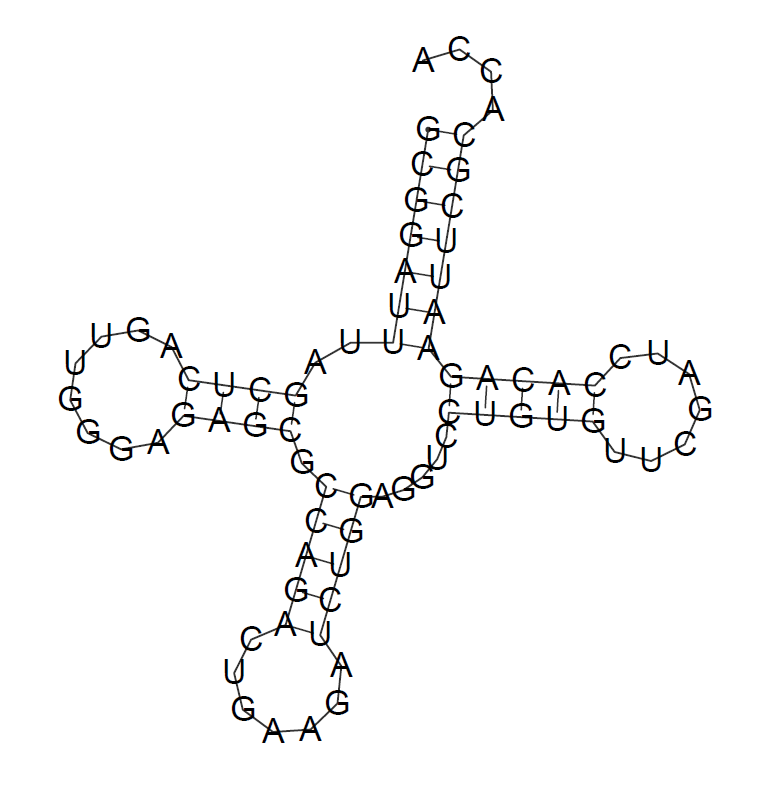
Взаимодействия нуклеиновых кислот
1. Предсказание вторичной структуры заданной тРНК
1.1. Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
При помощи программы einverted я нашел потенциальные стебли тРНК командой
einverted 1ehz.fasta -gap 7 -thr 10 -match 4 -mis -4 -outfile 1ehz.inv -outseq 1.fasta
Изменение настроек на меньший минимальный счет, уменьшение штрафов за гэпы и несовпадения и увеличение поощрения за совпадения так и не позволили найти больше инвертированных выравниваний.
Вторая выдача:
EMBOSS_001: Score 20: 5/5 (100%) matches, 0 gaps
49 ctgtg 53
|||||
65 gacac 61
очень похожа на T-стебель, и, как показано ниже, в сравнении с другими методами, скорее всего ей и является. Первая же выдача:
EMBOSS_001: Score 25: 10/12 ( 83%) matches, 1 gaps
22 gagcgccaga-ct 33
|| | ||||| ||
48 ctggaggtctaga 36
мало похожа на один стебель, но ее часть 5'-39-43-3' и 5'-27-31-3', скорее всего, является антикодоновым стеблем. D- и акцепторный стебли же мне этой программой найти не удалось.
1.2. Предсказание вторичной структуры тРНК по алгоритму Зукера
Алгоритм Зукера для последовательности данной тРНК был проведен при помощи последовательности команд:
export PATH=${PATH}:/home/preps/golovin/progs/bin
cat 11.fasta | RNAfold --noconv --MEA
ps2pdf rna.ps rna.pdf
Полученное изображение представлено на рисунке 1.
Эта схема вторичной структуры РНК, на мой взгляд, очень похожа на правду.
1.3. Сравнение методов предсказания вторичной структуры РНК
На основании всех полученных данных для сравнения использованных методов предсказания вторичной структуры РНК была составлена таблица 1.
| Участок структуры | Позиции по find_pair | Позиции по einverted | Позиции по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 5'-01-07-3' 5'-66-72-3' Всего 7 пар | - | 5'-01-07-3' 5'-66-72-3' Всего 7 пар |
| D-стебель | 5'-10-13-3' 5'-22-25-3' Всего 4 пары | - | 5'-10-13-3' 5'-22-25-3' Всего 4 пары |
| T-стебель | 5'-49-53-3' 5'-61-65-3' Всего 5 пар | 5'-49-53-3' 5'-61-65-3' Всего 5 пар | 5'-49-53-3' 5'-61-65-3' Всего 5 пар |
| Антикодоновый стебель | 5'-38-44-3' 5'-26-32-3' Всего 7 пар | 5'-39-43-3' 5'-27-31-3' Всего 5 пар | 5'-39-43-3' 5'-27-31-3' Всего 5 пар |
| Число канонических пар нуклеотидов | 21 | 10* | 21 |
*учитывать не стоит, так как из-за ненахождения всех стеблей не несет смысла.
По таблице видно, что наиболее эффективны и наиболее схожие результаты дали "find_pair" и алгоритм Зукера - первый метод лишь учел на одну неканоническую пару больше, которая, по моему мнению, в реальной РНК была бы недостаточно энергетически эффективна, так что в этом сравнении, как я думаю, наиболее точен алгоритм Зукера.
2. Поиск ДНК-белковых контактов в заданной структуре
В качестве структуры для поиска ДНК-белковых контактов мне был выдан комплекс 1d5y.
2.1. Скрипты JMol
С помощью команды define были заданы несколько групп атомов:
Это определение отображено в скрипте.
Также был написан скрипт, вызов которого в JMol даст последовательное изображение всей структуры, только ДНК в проволочной модели, той же модели, но с выделенными шариками каждого из заданных множеств атомов.
2.2. Поиск контактов каждого типа JMol
При помощи команд из скрипта была составлена таблица 2, в которой отражены ДНК-белковые контакты с цепью А белка из предложенной стуктуры.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 1 | 9 | 10 |
| остатками фосфорной кислоты | 10 | 9 | 19 |
| остатками азотистых оснований со стороны большой бороздки | 2 | 12 | 14 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
Как можно заметить, большинство контактов ДНК с белком в предложенной структуре неполярны, и только в остатках фосфорной кислоты число полярных взаимодействий примерно равно числу неполярных, да и общее число контактов больше - скорее всего, именно через фосфорные остатки и "заякориваются" белки в ДНК. Также можно заметить, что белок не образует контактов с малой бороздкой - скорее всего, она слишком тонка и в нее сложно встроиться аминокислотному остатку.
2.3. Популярная схема ДНК-белковых взаимодействий
С помощью программы nucplot взаимодействия были переведены в удобный для чтения человеком формат.
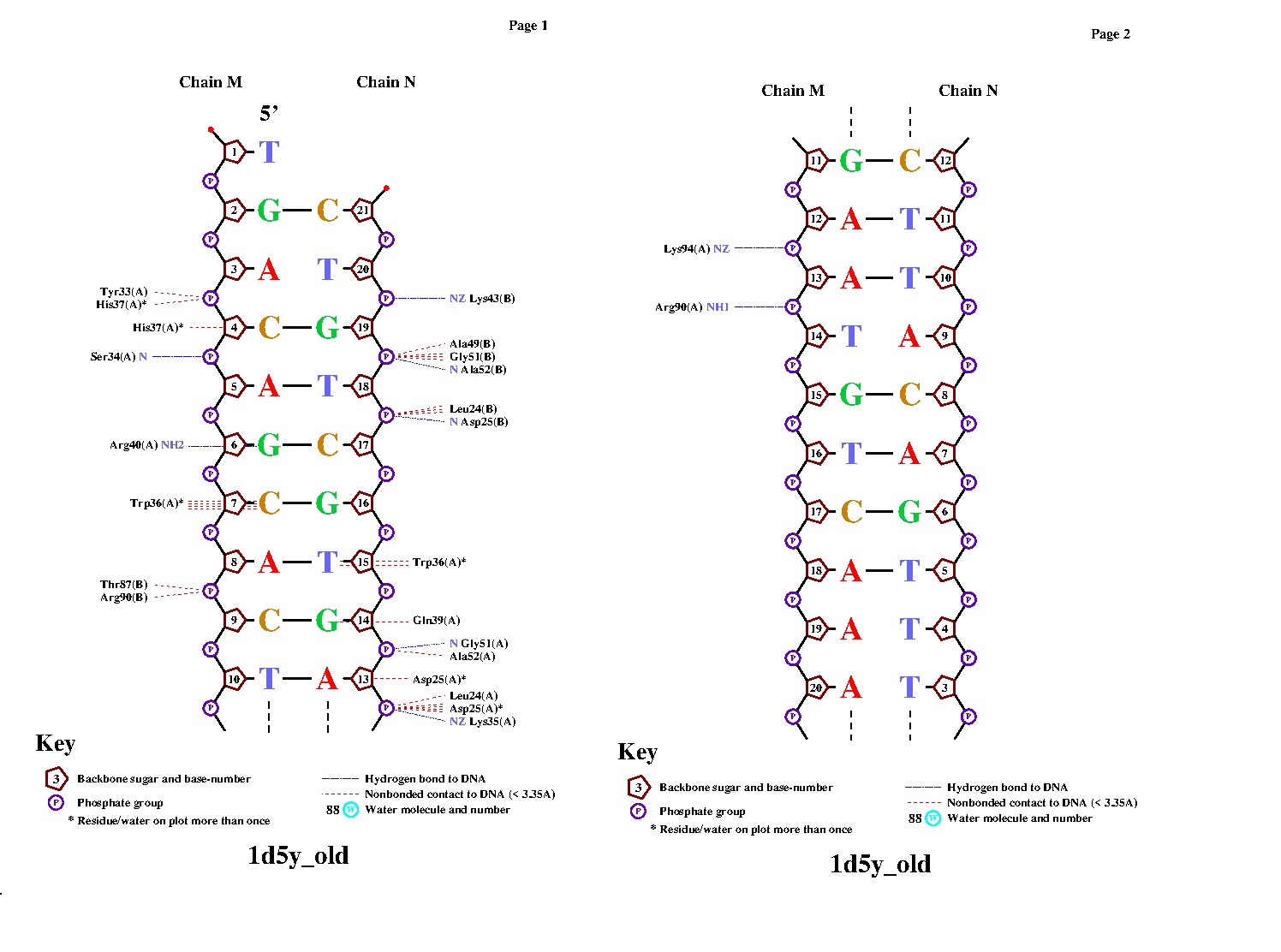
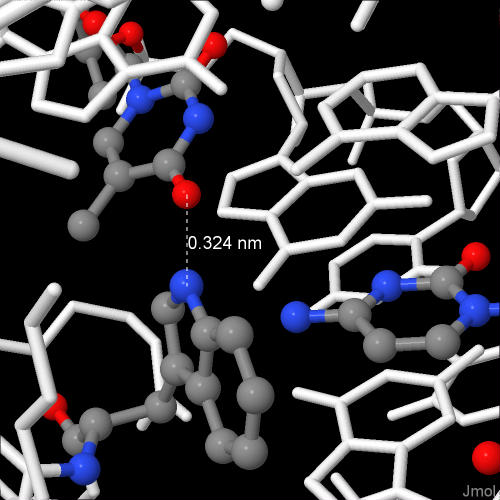
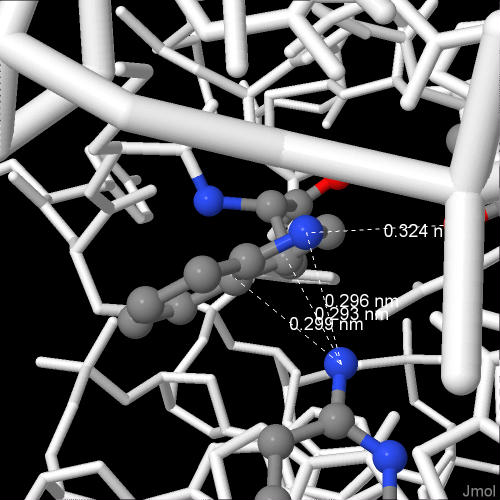
2.4. Местное взаимодействие ДНК с остатком аминокислоты
Судя по приведенной выше схеме, больше всего связей с ДНК имеет остаток Asp(25). Но, по моему мнению, наиболее важен для распознавания ДНК белком остаток Trp(36) - он связывается сразу с двумя азотистыми основаниями, единственным, по чему можно распознать ДНК.