Определение функции и таксономии нуклеотидной последовательности
В одной из предыдущих работ секвенированием по Сэнгеру была получена какая-то последовательность нуклеотидов. Что ж, пришло время выяснить, какую функцию она выполняет!
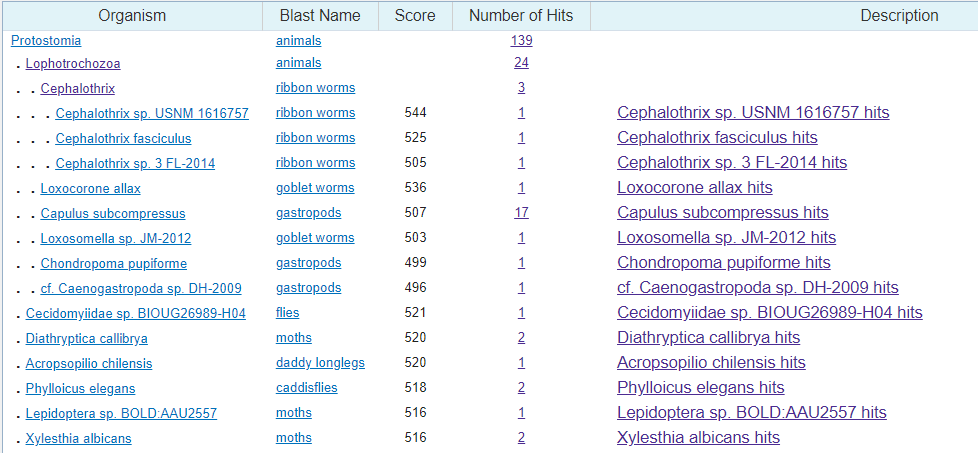
Для этого я воспользовался разновидностью алгоритма blastn - megablast, отличающейся оптимизацией для поиска очень близких гомологов, что необходимо для установления точной таксономии. В этот алгоритм я загрузил исследуемую последовательность, и оставил все настройки кроме длины слова неизменными. Длину слова я изменил на 32 для более скорого получения результата. В выдаче алгоритма прослеживается, что последовательность с наиболее высокой долей вероятности относится к таксону Lophotrochozoa(Рис. 1). Кроме того, из-за большого сходства вполне разумно предположить, что сама последовательность является геном COI и кодирует 1 субъединицу цитохром-C оксидазы - одного из ключевых ферментов кислородного метаболизма.
Затем все тем же алгоритмом, но с длиной слова 20 и только внутри таксона Lophotrochozoa было проведено еще одно выравнивание (выдача). Так я сузил круг поисков еще сильнее, до таксона Loxosomella, который в этот раз наиболее похож на искомый.
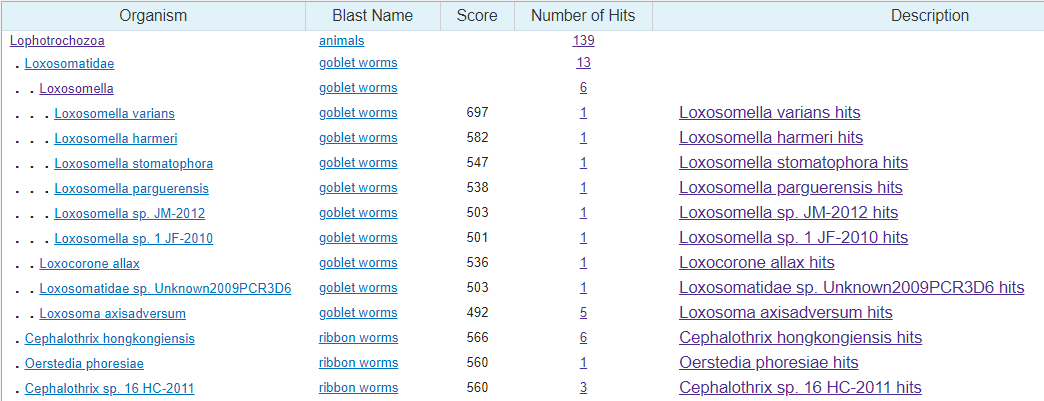
Уже внутри этого узкого таксона был запущен поиск с сохранением всех предыдущих настроек кроме длины слова (уменьшил до 16, благо поиск среди очень узкого круга последовательностей) и числа находок в выдаче (увеличил до 250 - чтобы наверняка найти наиболее узкий таксон. Находки представлены в выдаче, а самые похожие на родственные последовательности (имеющие минимальные e-value) проанализировал при помощи JalView, и, судя по выравниванию, хоть и искомая последовательноть наиболее близка Loxosomella varians, да и e-value BLAST с геном из этого организма стремится к нулю, но многие заменеы нуклеотидов относительно нее совпадают у анализируемой последовательности с одной из последовательностей Loxosomella harmeri или Loxosomella stomatophora, так что я предроложу, что точнее рода - Loxosomella sp. - определить таксон по полученной последовательности не представляется возможным.
Поиск генов белков в неаннотированной нуклеотидной последовательности
Для упражнения я выбрал один из контигов рассмотренной в прошлой работе - scaffold0498. Его в виде fasta-последовательности я подал на вход blastx - лучшему варианту нуклеотидного бласта при необходимости найти информацию о транслируемом с последовательности белке. Из настроек - все по дефолту кроме целевого таксона (я выбрал линяющих для ускорения процесса поиска), длины слова (выбрал 3 для большей вероятности получить искомое), expect value и числа находок (0,1 и 250 соответственно по тем же причинам). В выдаче можно проследить совпадения отдельных частей транскриипта данной последовательности с известными белками, что наглядно показано на рис. 3.

С довольно большим счетом в транскрипте скэффолда видно два потенциальных белка - разветвленноцепочечная аминокислототрансфераза (первая салатовая колонка примерно до середины рисунка 3) и гепаринсульфат О-сульфотрансфераза (вторая салатовая колонка во всю высоту рисунка 3, продолжается и дальше). Я предпочту найти далее границы гена второго из перечисленных белков, так как с ним больше совпадений.
Судя по выравниваниям в blast, искомый ген находится в пределах 19608-19913bp в зависимости от выравнивания и содержит менее трети белковой последовательности (под 100 аминокислот в лучшем случае из более 300 аминокислот полного белка). Смею предположить, что это может быть связано с тем, что следующая за 19913 нуклеотидом область (а последжовательность, которую я загрузил, похоже, является обратной к реальным) является интроном гена, а остальные экзоны оказались за пределами скэффолда.
Интепретация карты локального сходства гомологичных хромосом двух бактерий
Для построения карты локального сходства были взяты полные геномы двух относительно близких видов бактерий - Bacillus subtilis и Bacillus velezensis. Эти геномы были загружены в качестве входных данных для парного выравнивания в megablast, выбранного в данном случае как наиболее эффективный алгоритм для очень схожих последовательностей. Полученная карта локального сходства представлена на рисунке 4. Карта получена при стандартных настройках алгоритма, чище от их изменения без потери иннформации не становилось.
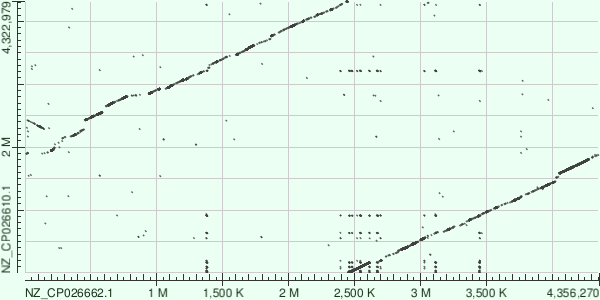
Очевидно, что разрыв посередине является следствием того, что у данных бактерий хромосомы кольцевые - при секвенировании начинали с разных точек и это вряд ли мутация. Зато мутация хорошо видна на отрезке 2300К-2450Кbp по горизонатльной оси - это явно инверсия с транслокацией. На отрезке 1450К-1550Кbp по той же оси хорошо видна делеция части генома у Bacillus subtilis, а на отрезке 800К-950Кbp по вертикальной оси - делеция у Bacillus velezensis