11. Введение в анализ данных NGS
Задачей этой части работы стало подготовление необходимых файлов, изучение качества их чтений, фильтрование чтений и подготовка их к картированию.
Итак, мне выпала сборка 15 хромосомы.
Описание файла с референсом
Файл с референсом очень похож на обычную Fastsa-последовательность ДНК. В самом начале идет огромный массив неопределенных нуклеотидов, что, похоже, соответствует последовательности теломераз или просто проблемами с секвенированием концевых участков. Примечательно, что в конце файла также есть большой N-повтор, который, однако, в десятки, если не сотни или тысячи раз короче массива в начале. Смысловая часть начинается только после 17000000 позиции!
Индексация референса
Для сборки по референсу ему нужно бы быть проиндексированным. Для индексации была использована утилита BWA командой:
bwa index -a bwtsw chr15.fna &> log_bwa
"index" тут было использовано с целью индексации референса, "-a bwtsw" позволяет работать по алгоритму, оптимизированному для генома человека. Вывод программы можно посмотреть тут.
В результате работы BWA при поданной на вход последовательности хромосомы были получены несколько файлов chr15.fna, где:
Описание образца
Мне достались парноконцевые чтения полноэкзомного секвенирования SRR10720419 Homo sapiens, выполненные на "Illumina Genome Analyzer IIx" с 41.3M чтений.
Проверка качества исходных чтений
Качество исходных чтений оценивалось при помощи программы fastqc по командам:
fastqc SRR10720419_1.fastq.gz &> log_fastqc1, вывод тут
fastqc SRR10720419_2.fastq.gz &> log_fastqc2, вывод тут
Среди полученных файлов легче всего осмыслить html-файлы - посмотреть их можно тут: SRR10720419_1 и SRR10720419_2.
Легко заметить, что прямых и обратных чтений плучилось поровну - по 41277367.
Per base sequence quality
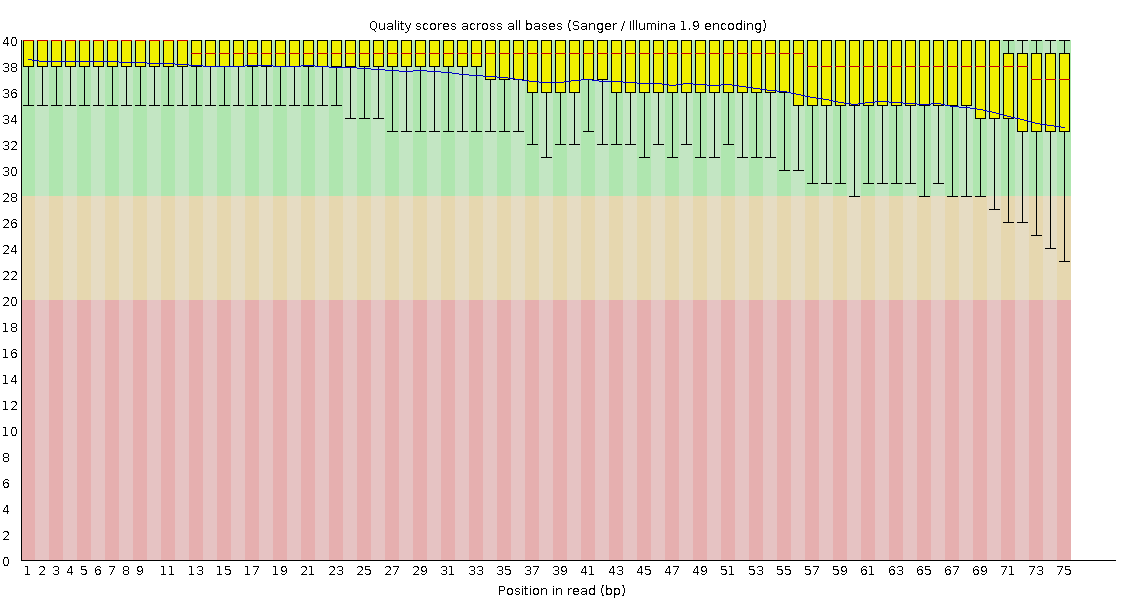
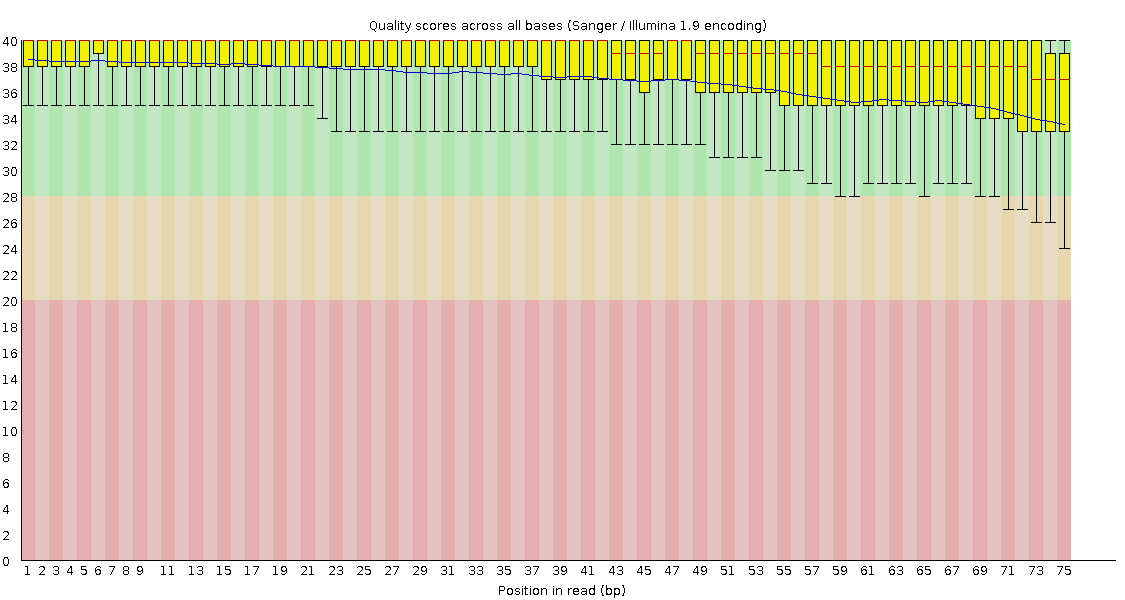
По рис. 3, 4 можно оценить качество чтений как очень хорошее по всей длине, и лишь под конец некоторая часть из них имеет качество ниже 28, причем на прямых чтениях качество в среднем чуть хуже, чем на обратных.
Per sequence quality scores
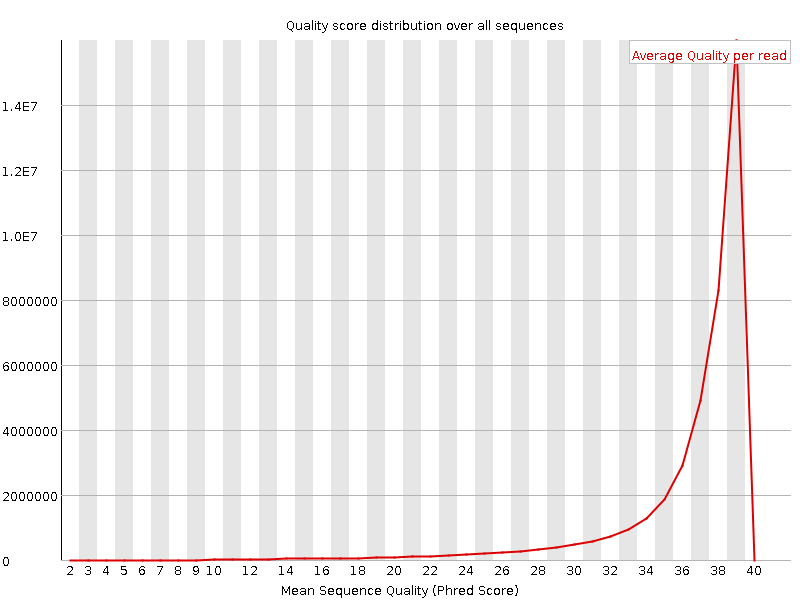
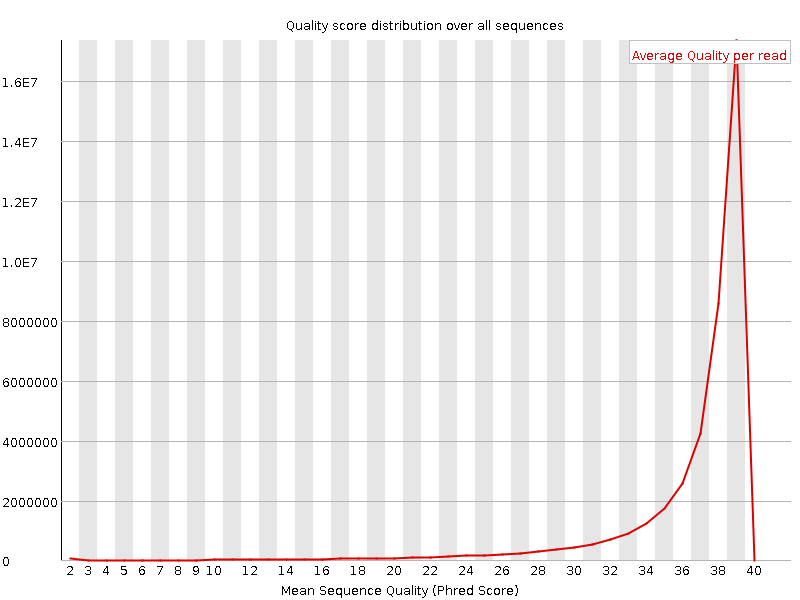
По рис. 5, 6 видно, что подавляющее большинство как прямых, так и обратных чтений имеют качество 39, а значения качества ниже 28 показывает крайне малая доля чтений, что также говорит о крайне высоком качестве секвенирования. И опять для обратных чтений доля лучших чуть выше, чем у прямых.
Sequence Length Distribution
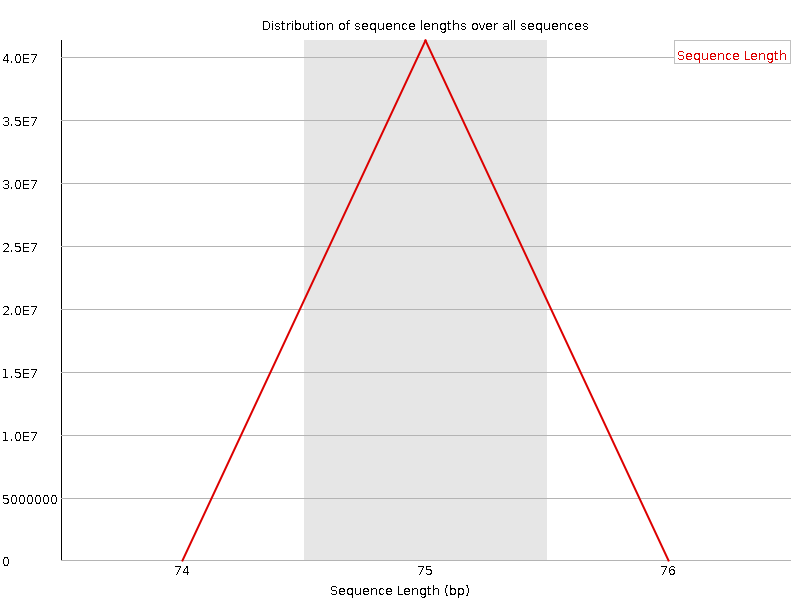
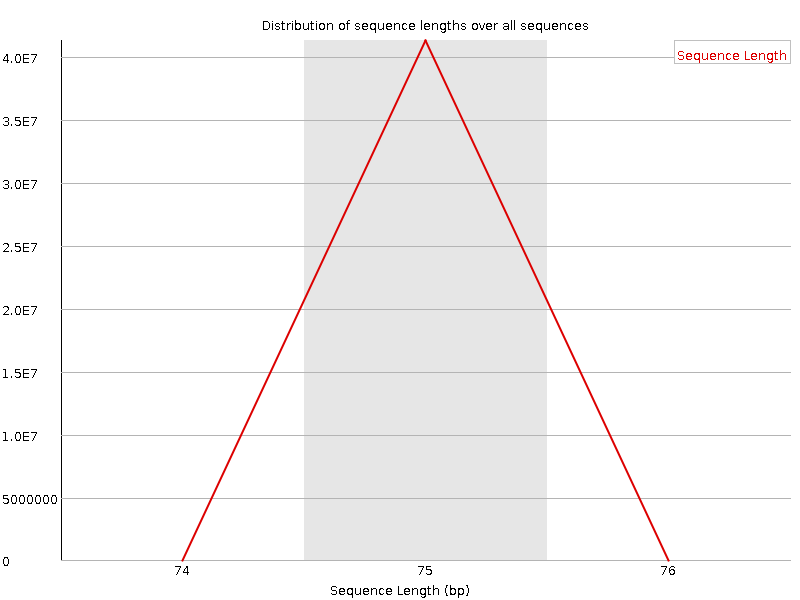
В первую очередь для рис. 7, 8 хочется отметить их идентичность - прямые и обратные чтения имеют одинаковое распределение длин, что говорит об очень хорошей стабильности выбранного метода секвенирования. В итоге ВСЕ чтения имеют длину ровно 75 bp.
Фильтрация чтений
Несмотря на очень хорошее качество чтений, я все же предпочел отсечь некачественные крохи при помощи программы trimmomatic командой
java -jar /usr/share/java/trimmomatic.jar PE -threads 15 -phred33 SRR10720419_1.fastq.gz SRR10720419_2.fastq.gz output_SRR10720419_1_paired.fastq.gz output_SRR10720419_1_unpaired.fastq.gz output_SRR10720419_2_paired.fastq.gz output_SRR10720419_2_unpaired.fastq.gz TRAILING:20 MINLEN:50 &> log_trimmomatic, вывод тут
Тут "РЕ" сигнализирует о парных концах, "-threads 15" ускоряет выполнение за счет использования 15 ядер, "-phred33" задает формат scorе, "TRAILING:20" удаляет нуклеотиды с конца чтений с качеством ниже 20 и "MINLEN:50" отфильтровывает чтения короче 50 bp. "SRR10720419_1.fastq.gz" и "SRR10720419_2.fastq.gz" - входные файлы, остальные файлы выходные.
Проверка качества триммированных чтений
Для контроля качества триммирования полученные 4 файла были опять прогнаны через fastqc:
fastqc output_SRR10720419_1_paired.fastq.gz &> log_fastqc1p, вывод тут, html тут.
fastqc output_SRR10720419_1_unpaired.fastq.gz &> log_fastqc1u, вывод тут, html тут.
fastqc output_SRR10720419_2_paired.fastq.gz &> log_fastqc2p, вывод тут, html тут.
fastqc output_SRR10720419_2_unpaired.fastq.gz &> log_fastqc2u, вывод тут, html тут.
Судя по записям, полученным от fastqc, в обоих наборх осталось по 39759554 bp (96,32% от начального числа bp), что еще раз подтвержает в целом высокое качество начальных данных.
Per base sequence quality
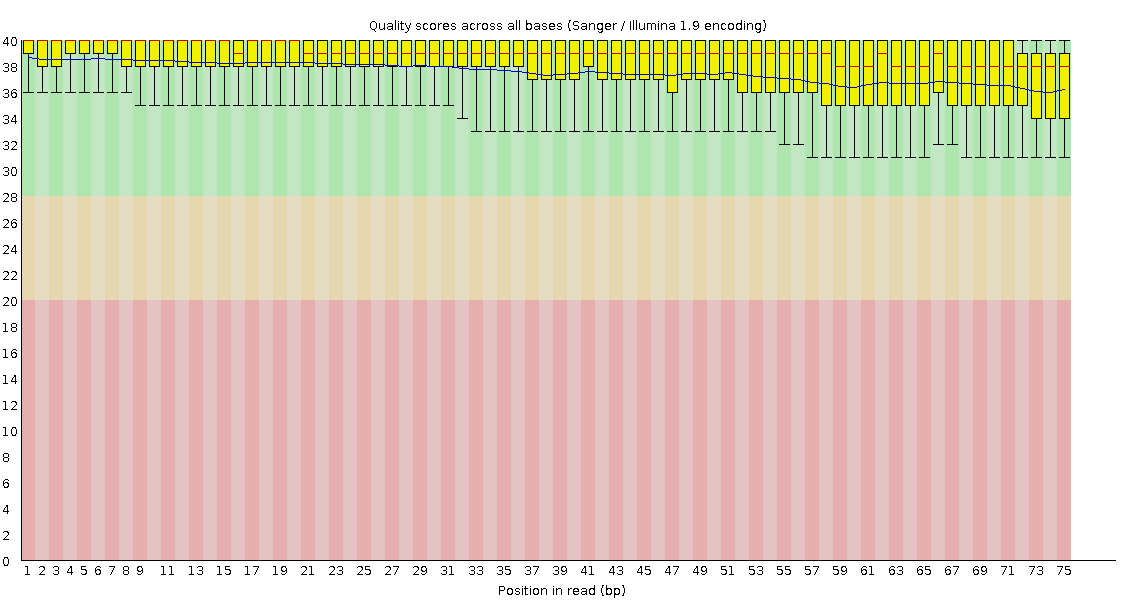
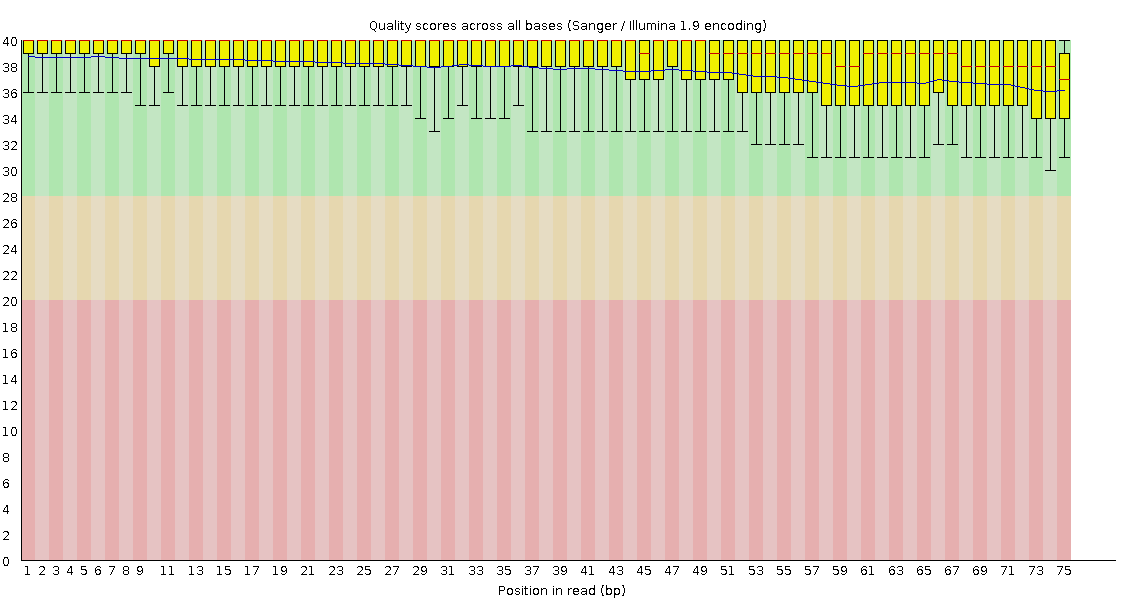
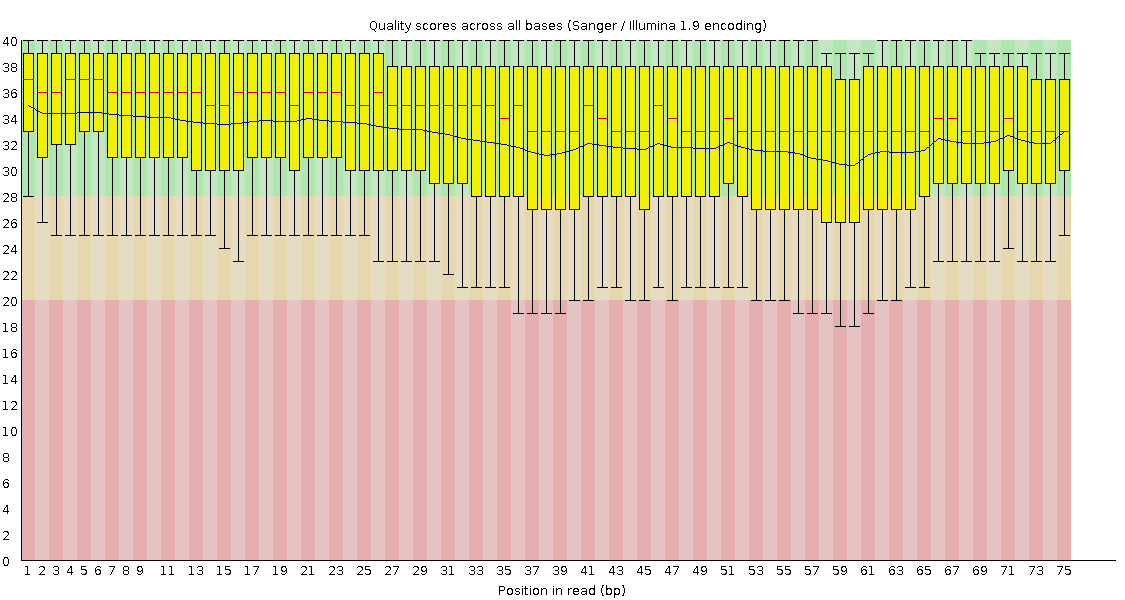
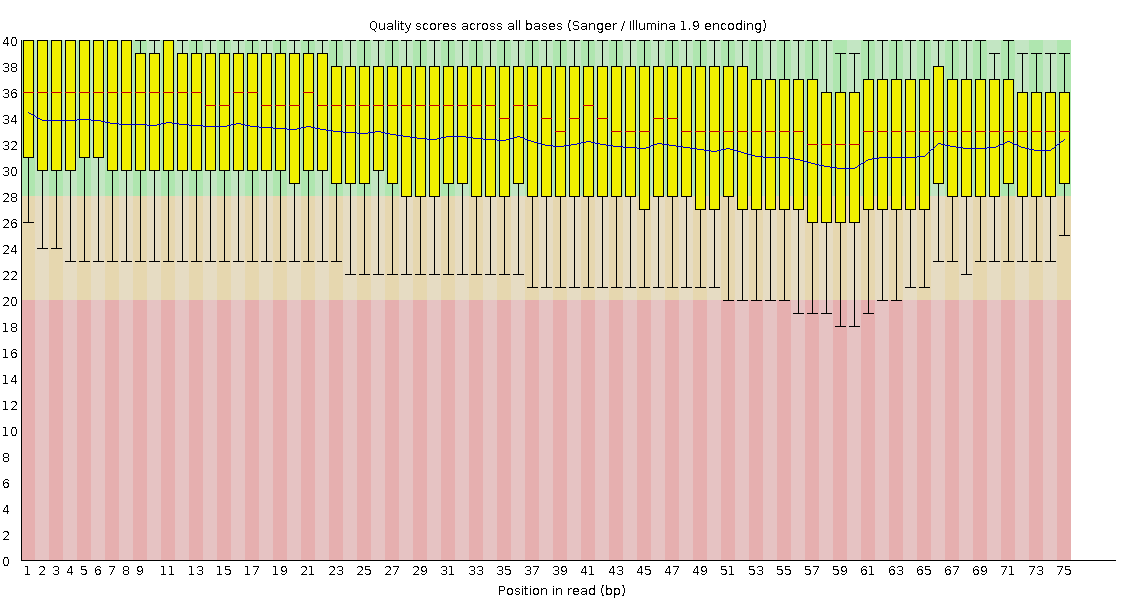
По рис. 9, 10, 11, 12 Становится очевидно, что триммирование чтений прошло успешно: все низкокачественные риды оказались в файлах unpaired, в то время как нужные нам целевые высококачественные со счетом не ниже 30 (уже виден выигрыш над изначальным состоянием) сохранились в отдельных файлах.
Per sequence quality scores
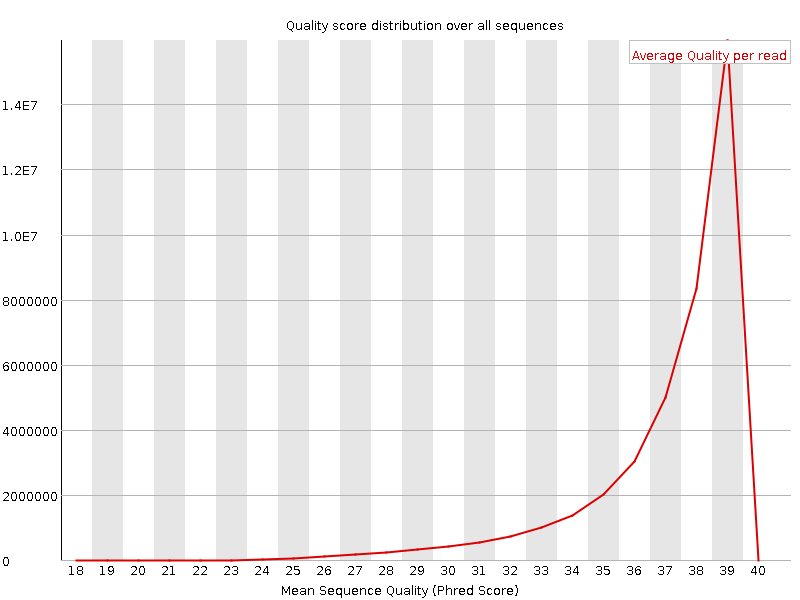
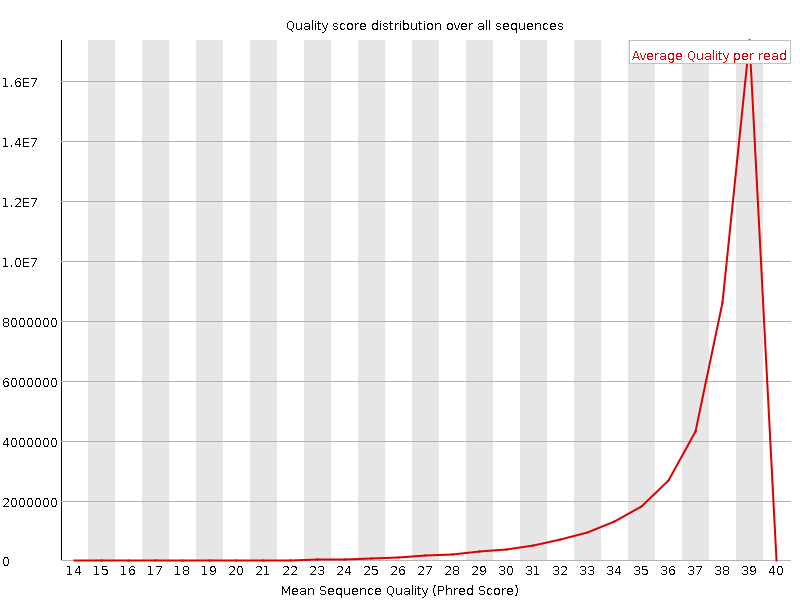
По рис. 13, 14 и в сравнении с рис. 5, 6 видна лишь одна разница - отсчет таблицы начинается с 14 для обратного и с 18 для прямого прочтения, что связано с удалением коротких прочтений.
Sequence Length Distribution
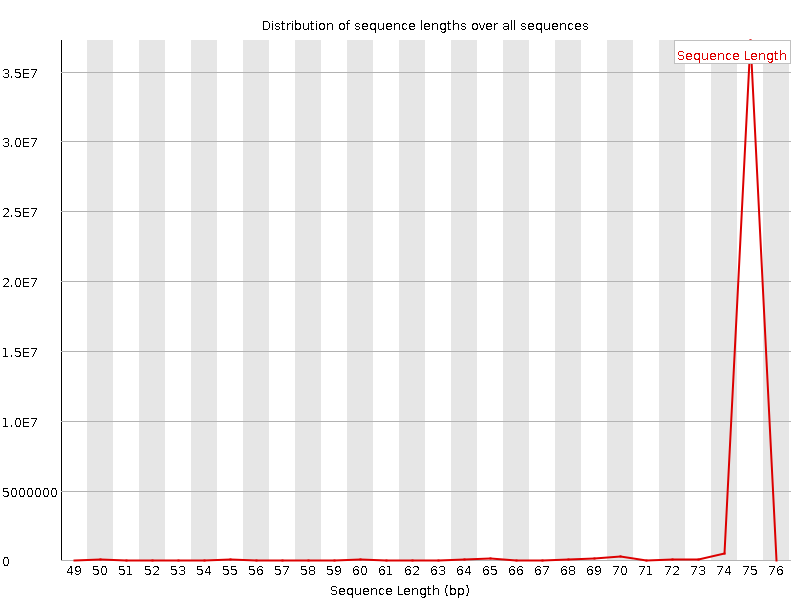
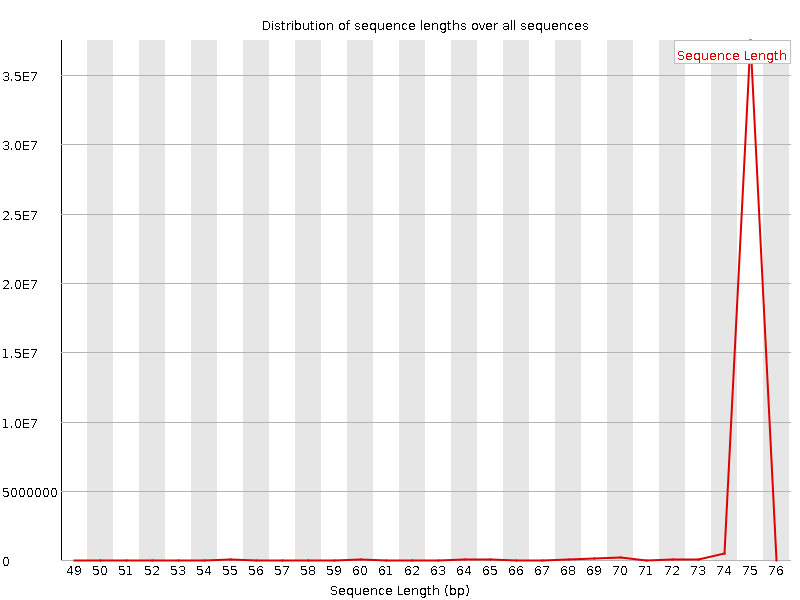
И опять рис. 15, 16, как и рис. 7, 8, почти идентичны и отличаются лишь небольшим сдвигом по масштабу. И снова подавляющее большинство составляют чтения длины 75, но появились и более короткие - обрезанные при триммировании.
12. Ресеквенирование. Поиск полиморфизмов у человека.
Теперь нужно картировать триммированные чтения на геном человека и подготовить файлы для получения списка вариантов полиморфизмов.
Картирование чтений на референсный геном
Картирование проведено программой bwa mem с получением файла с расширением .sam командой
bwa mem -t 15 chr15.fna output_SRR10720419_1_paired.fastq.gz output_SRR10720419_2_paired.fastq.gz > mapped.sam 2> log_bwa
В данной команде "bwa mem" - собственно, сама программа, "-t 15" - распараллеливание для нетерпеливых на 15 ядер, "chr15.fna" - референс, "output_SRR10720419_1_paired.fastq.gz" и "output_SRR10720419_2_paired.fastq.gz" - сами картируемые файлы, полученные в предыдущей части задания, "mapped.sam" - файл с картированием и "log_bwa" - запись со стандартного потока ошибок.
Анализ sam файла с выравниванием
Этот файл состоит из шапки и собственно основной таблицы (рис. 17). Шапка выделена значком "@" и содержит информацию о программе и ее настройках.
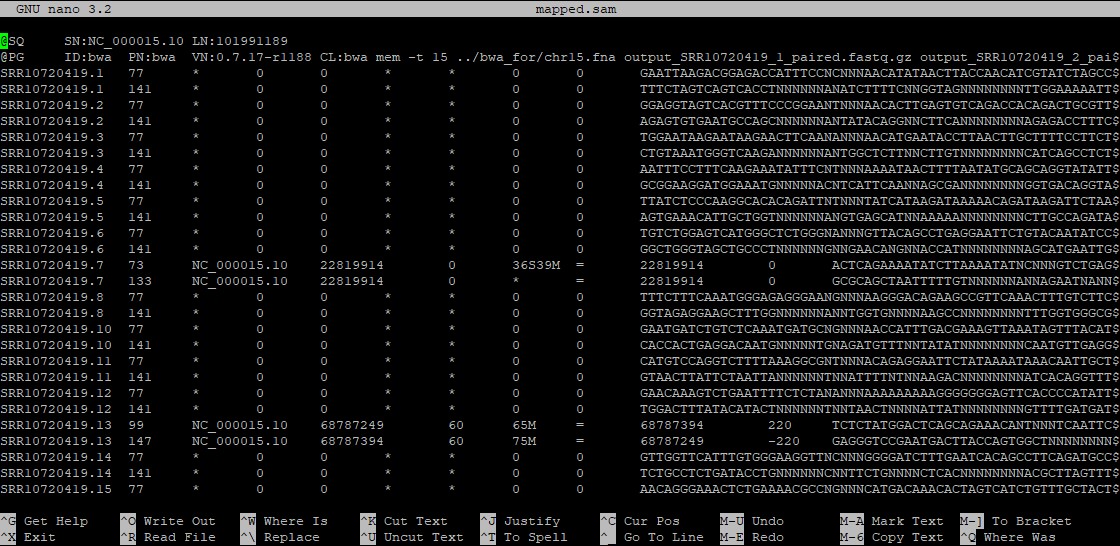
В таблице же содержатся данные о картировании ридов. Подробно значение каждого столбца представлено в таблице 1.
| Название | Описание | Тип переменной |
|---|---|---|
| QNAME | Имя чтения | String |
| FLAG | Комбинация подразрядных FLAGов | Int |
| RNAME | Имя референса | String |
| POS | Позиция в референсе, на которую картировался первый нуклеотид | Int |
| MAPQ | Качество картирования* | Int |
| CIGAR | Краткий отчет о выравнивании | String |
| RNEXT | RNAME следующего чтения** | String |
| PNEXT | POS следующего чтения | Int |
| TLEN | Длина картирования | Int |
| SEQ | Последовательность чтения | String |
| QUAL | Phred-score каждого нуклеотида в риде | Int |
| AS | Cчет выравнивания по aligner | String |
*равно -10 log10 Pr{позиция картирования неверная} и округляется до целых
**если значение "=", то соответствует RNAME
Получение и индексирование bam файла
Но весит такой sam-файл аж 15,73ГБ! Чтобы облегчить жизнь используемому нами узлу, этот файл был переконвертирован в сортированный bam-файл и удален.
samtools sort -@ 15 -o mapped.bam mapped.sam
Где "samtools sort" - сама программа, "-@ 15" обеспечивает скорость за счет распараллеливания на 15 ядер, "-o" задает выходной файл а "mapped.bam" и "mapped.sam" - сами выходной и входной файлы. Такой бинарный файл намного компактнее - он занял всего 4,58ГБ!
Далее bam-файл нужно проиндексировать, для этого я воспользовался командой
samtools index -@ 15 mapped.bam
Анализ bam файла
Чтобы проанализировать бинарный файл, его нужно как-то привести в читаемый вид. Помочь с этим может команда:
samtools flagstat -@ 15 mapped.bam > flag.txt
Из выданного файла можно получить следующую информацию: картировано 8386874 чтений (10,54%), в корректно картированных парах 6373786 чтений (8,02%). Отличие этих чисел можно объяснить например тем, что одно из чтений может картироваться в несколько мест генома.
Получение картированных чтений
Из-за того, что мы картируем полный экзом на только лишь одну хромосому, ожидаемо возникает множество некартированных последовательностей, от которых было бы неплохо избавиться. Чтобы получить название выстраиваемой хромосомы, я воспользовался командой:
samtools faidx ../bwa_for/chr15.fna
Эта команда выдала следующе:
NC_000015.10 101991189 70 80 81
Используя эти данные был получен новый файл с отсечением лишних чтений командой:
samtools view -@ 15 -h mapped.bam NC_000015.10 > chr15.sam
Полученный файл уже не имеет строк с нулевым картированием. Опять же, для компактности он был переведен в бинарный формат и удален.
samtools sort -@ 15 -o chr15.bam chr15.sam
И опять полученный файл был проанализирован при помощи команды:
samtools flagstat -@ 15 chr15.bam > flagch.txt
В выданном файле уже картировано 87,20% чтений (против 10,54% в прошлый раз), а в корректно картированных парах 66,54% чтений (против 8,02% до этого) с сохранением абсолютных значений - 8386874 и 6373786 соответственно. Что ж, пора извлечь только корректно картированные чтения! Для этого я воспользовался командой:
samtools view -@ 15 -f 0x2 -bS chr15.bam > correct_chr15.bam
И уже который раз файл проанализирован программой flagstat:
samtools flagstat -@ 15 correct_chr15.bam > flagcor.txt
В выданном файле, как и ожидалось, все корректно картированные пары чтений (6373786) были взяты из корректно картированных пар чтений предыдущей версии файла, а просто картированных чтений оказалось чуть больше - 6375175 и составили вполне ожидаемые 100%, такое преобладание просто картированных чтений над корректно картированными снова соответствует предыдущим файлам.
Под конец этот уже почти идеальный файл был проиндексирован:
samtools index -@ 15 correct_chr15.bam
Подготовка выравнивания к поиску вариантов
При подготовке к поиску вариантов требуется промаркировать дублированные чтения с помощью команды picard MarkDuplicates:
picard MarkDuplicates -M metrix.file.txt -I correct_chr15.bam -O marked.bam
К сожалению, занять больше ядер не удалось, и процесс шел долго...
К полученному файлу опять применена команда flagstat:
samtools flagstat -@ 15 marked.bam > flagmark.txt
В выданном файле содержится информация о числе дублированных чтениий - их 459564.
13. Продолжение
Наконец пришла пора искать варианты!
Получение вариантов
Для получения вариантов использовалась программа bcftools:
bcftools mpileup --threads 15 -f ../bwa_for/chr15.fna marked.bam | bcftools call -mv -o chr15.vcf
Тут "bcftools mpileup" - собстенно алгоритм, "--threads 15" - команда на использование множества ядер ради скорости работы, "-f" намекает на индексированность референса, "../bwa_for/chr15.fna" - путь к референсу, "marked.bam" - сам файл с обработанными ранее чтениями, "bcftools call" - командда для вызова вариантов, "-mv" задает вызов редких и многоаллельных вариантов с записью в файл только вариантов, а "-o chr15.vcf" - опция выходного файла и его название.
Полученный vcf-файл, как и sam-файл, состоит из шапки и таблицы (рис. 18). Только у свежеполученного файла шапка гораздо больше - там содержатся не только настройки его создания, но и, насколько я понял, информация об общем количестве различных типов отличий от референса, а также названия колонок таблицы.
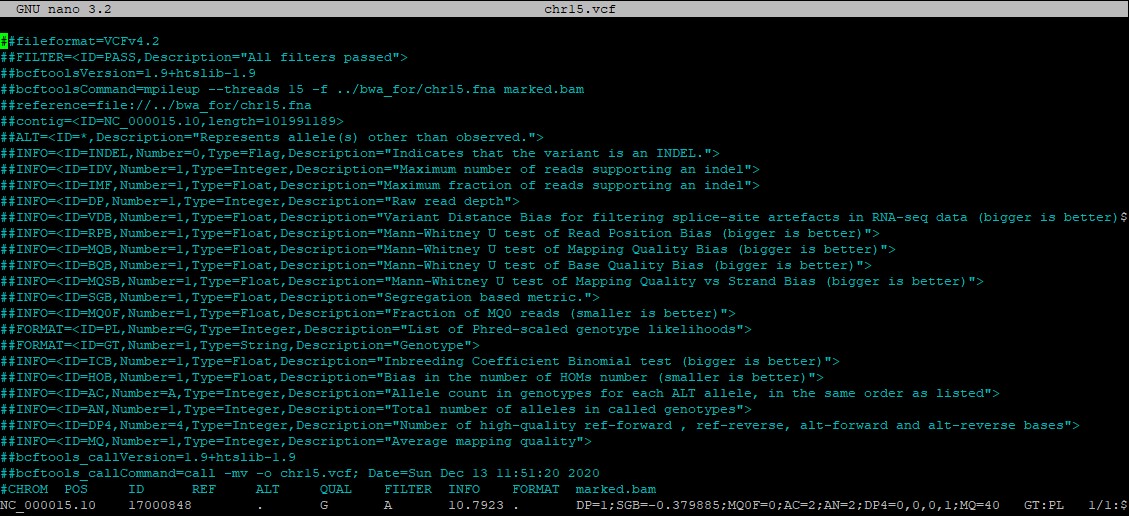
В самой же таблице деcять колонок с информацией о вариантах (рис. 19):
Далее этот файл можно проанализировать командой:
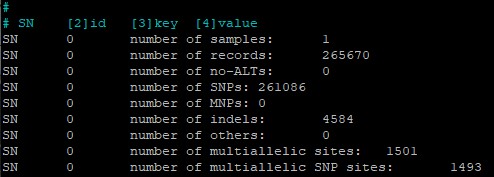
bcftools stats chr15.vcf > chr15stat.txt
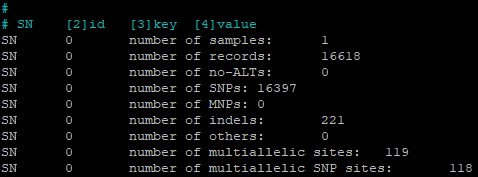
Судя по данным в этом файле (рис. 20), всего в данной мне для сборки хромосоме 265670 вариантов, почти все из них - 261086 - являются однонуклеотидными заменами (SNP), а оставшиеся 4584 вариантов - индели.
Фильтрация вариантов
Для большего доверия полученным вариантам отфильтруем варианты с качеством выше 30 и покрытием больше 50 командой:
bcftools filter -i'%QUAL>30 && DP>50' --threads 15 chr15.vcf -o chr15filtr.vcf
И полуенный файл опять же приводится к читаемому виду командой:
bcftools stats chr15filtr.vcf > chr15filtrstat.txt
И в отфильтрованном файле (рис. 21) оказалось уже намного меньше вариантов - 16618, из которых снова почти все - SNP (16397 случаев), а инделей уже 221. Интересно, что фильтрацией отброшена гораздо большая доля инделей, чем SNP.
Изучение покрытия
Что ж, теперь мы столкнулись с новой проблемой - мы не знаем нашего таргета. Предложено поступить так: найти покрытые участки, отобрать участки с хорошим покрытием, такие участки считать таргетом, проанализировать именно их. Для начала получим участки генома, покрытые предложенными чтениями хотя бы один раз командой:
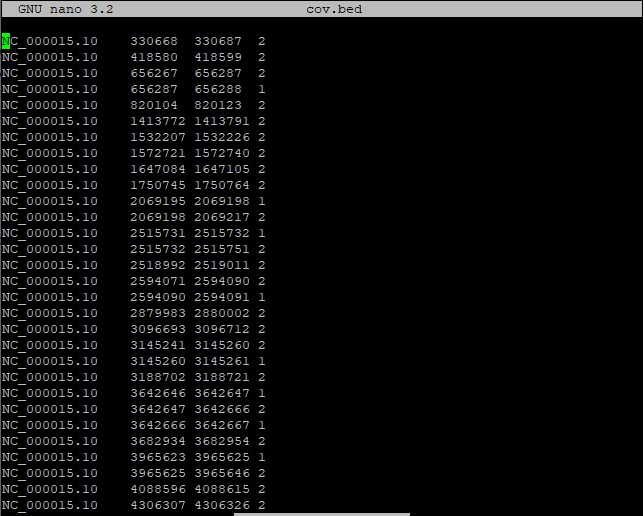
bedtools genomecov -bg -ibam marked.bam > cov.bed
Полученный файл (рис. 22) выглядит достаточно просто: в четырех столбцах описаны название референса, начало варианта, конец варианта и его покрытие соответственно.
После этого отберем только покрытые более 50 раз участки командой:
awk '$4 > 50' cov.bed > cov50.bed
В этом файле оказалось 1215992 (25,24% от нефильтрованного файла) строк, что было подсчитано командой wc:
wc -l cov50.bed
Похоже, это и есть наши таргеты.