Поиск последовательности Шайн-Дальгарно
Использовался геном бактерии Streptomyces fodineus.
1. Подготовка входных последовательностей
Скрипт для подготовки предоставлен Д. Звездиным. Представляю его ниже с небольшими изменениями.
from Bio import SeqIO
import pandas as pd
import numpy as np
from IPython.display import Image
with open("sequence.gb", "r") as file:
table = SeqIO.read(file, "gb")
coordinates = []
with open("coordinates.tsv", "w") as file_write:
for feature in table.features[1:]:
start, end, strand = int(feature.location.start), int(feature.location.end), feature.location.strand
try:
locus_tag = feature.qualifiers["locus_tag"][0]
except KeyError:
locus_tag = f"unknown_locus_tag_{start}_{end}"
if (feature.type == "CDS") and ("pseudo" not in feature.qualifiers.keys()) and (end - start > 300) and
(feature.qualifiers["product"][0] != "hypothetical potein"):
coordinates.append("\t".join([str(start), str(end), str(strand), locus_tag]))
print(*coordinates, sep="\n", file=file_write)С помощью скрипта были отобраны хорошо аннотированные (не гипотетические) кодирующие последовательности длиной более 300 п.н. и получены их координаты.
coordinates_table = pd.read_csv("coordinates.tsv", sep="\t", names=["start", "end", "strand", "name"])
with open("Streptomyces_fodineus.fasta", "r") as fasta_file:
fasta = SeqIO.read(fasta_file, "fasta")
clear_sequences = []
with open("clear_sequences.fasta", "w") as file_write:
for i in range(3000):
name = coordinates_table.loc[i, "name"]
if str(coordinates_table.loc[i, "strand"]) == "1":
start = int(coordinates_table.loc[i, "start"])
sequence = str(fasta.seq[start - 30: start])
else:
start = int(coordinates_table.loc[i, "end"])
sequence = str(fasta.seq[start: start + 30].reverse_complement())
clear_sequences.append(f">{name}")
clear_sequences.append(sequence)
print(*clear_sequences, sep="\n", file=file_write)
Далее из fasta файла с хромосомой по полученным координатам были скачаны 3000 последовательностей с 30 нуклеотидами upstream генов: 100 последовательностей для MEME и 1000 для FIMO.
for feature in table.features[1:]:
try:
if ("RNA" in feature.type) and ("16S" in feature.qualifiers["product"][0]):
print(feature.location)
except KeyError:
pass
print(str(fasta.seq[8019814:8019844].complement()))Также в файл с данными для обучения был добавлен 3' концевой участок 16S РНК CGCCGACCTAGTGGAGGAAA.
Файл с последовательностями для MEME.
Файл с последовательностями для FIMO.
2. Поиск мотивов с помощью MEME
Был использован сервис MEME Suit.
Параметры запуска:
meme meme.fasta -dna -oc . -nostatus -time 14400 -mod zoops -nmotifs 1 -minw 9 -maxw 9 -objfun classic -markov_order 0В результате работы программы был найден мотив, содержащий последовательность GGAGG, похожий на последовательность Шайн-Дальгарно (Рис.1). Он встретился в 37 последовательностях из 101. В силу того, что сигнал посадки рибосомы может находиться на разном расстоянии от сайта трансляции, некоторые позиции могут быть неоднозначны.
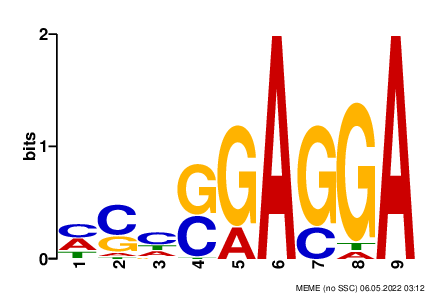
3. Поиск SD в выборке для тестирования с помошью FIMO
Параметры запуска:
fimo --oc . --verbosity 1 --thresh 0.01 --norc motifs.meme fimo.fastaПри уровне значимости 0,01 мотив был найден в 629 последовательностях из 1000.