BLAST.
Поиск гипотетических гомологов белка TRMB_BACSU в разных банках.
Для поиска гипотетических гомологов белка TRMB_BACSU воспользуемся алгоритмом BLAST. Результаты поиска приведены в "Таблице 1".
Таблица 1. Результаты поиска гипотетических гомологов белка TRMB_BACSU в разных банках.

Как видно из таблицы нам удалось найти гомологи белка TRMB_BACSU по базе данных Swiss-Prot и "nr"("Non-redundant protein sequences"). Помимо этого из третьего столбца видно, что существуют и PDB структуры гомологов белка. Возникает вопрос: "почему разное число находок?". Больше всех находок по базе Non-redundant protein sequences , что впринцепе логично, поскольку поиск через нее идет сразу на нескольким базама данных(SwissProt, PIR, и PDB) и и в него попадают все возможные по степени изучености белки. PDB дает маленькое количество находок, поскольку структура не всех белков установленна. Самый, на мой взгялд, адекватный результат дает поиск по SwissProt, т.к. там хранится информации о хорошо изученных и проверенных белках.
(использованные параметры BLAST: E-value < 1e-10; E-value < 1 с предельным размером выдачи - 500)
Поиск гипотетических гомологов белка TRMB_BACSU с фильтром по таксонам.
Таблица 2. Результаты поиска гипотетических гомологов белка TRMB_BACSU с фильтром по таксонам.
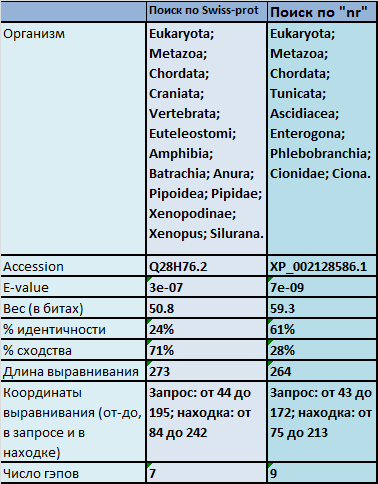
С помощью прогрмаммы BLAST, можно вести поиск гомологов белка по таксонам в других систематических группах.Основной задачей было найти удовлетворительный гомолог (критерий: e-value < 1e-4) начельного белка в наиболее "далеком" таксоне. Результаты поиска представленны в Танбице 2.
Парное выравнивание TRMB_BACSU(O34522) и его гипотетического гомолога (Q518Y2) с помощью BLAST.
Рис. 1. Результаты выравнивания при e-value = 10(стандартное значение критерия).
.png)
Праграмма BLAST позволяет сделать выравнивание парное начального белка с его гипотетическим гомолого( галочка у Align two or more sequences и появится новое окошко для ввода). Она строит строит карту локального сходства(Рис. 1, Рис. 2), на которой по бокам находятся иследуемые белки. График(прямая) образуется только в местах полного совпадения последовательностей.
Рис 2. Результаты выравнивания при e-value = 0,01.

Из Рис. 1 и Рис. 2 можно сделать вывод, что значение параметра e-value не оказывает никого влияния на сходство последоватльностей.