Парные выравнивания белков. Применение алгоритмов парных выравниваний к белку TRMB_BACSU.
Сравнение матриц аминокислотных замен.
Матрица аминокислотных замен - это матрица, в которой по стороке и столбцу , в единственном порядке, расположены 20 незаменимых аминокислот. В ij месте пересечения столбца и строки находится число. Это число - вес аминокислотной замены, показывающий примерную оценку замен одной аминокислоты на другую. Оно получается сходя из статистических данных базы BLOKS о всречаемости замени и возможности сдвига(замен на геп) в биологических организмах.
Матрица BLOSUM не является приминимой для всех белков, в соновном только для цитоплазматических (т.к. в BLOKS собранны в основмном цитоплазматические). Для мембранных белков существуют специальные матрицы такие как: PHAT (predicted hydrophobic and transmembrane). Как ни старнно, но различия в замене аминокислот между PHAT и BLOSUM заменото хорошо прослеживают(что дает повод смотреть к каким группам относиться анализируемый белок).
За долгое время информация базы данных BLOKS не раз успела обновиться. После реконструкции BLOSUM62 по последним данным из BLOKS, это особо хорошо прослеживается.
Таблица 2. Сравнение весов замен аминокислоты глутамата (Glu, E)
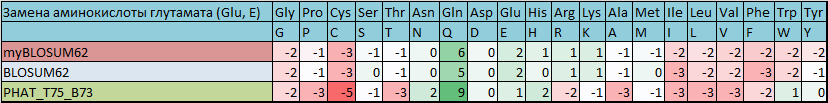
В первой колонке: красным выделина матрица BLOSUM62 просторееная на основе последних данных BLOKS; серым - старая матрица BLOSUN62; зеленоватым - PHAT(матрица для мембранных белков)
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана.
Алгоритм Нидлмана — Вунша является примером динамического программирования. Это алгоритм для выполнения выравнивания двух последовательностей, который используется в биоинформатике при построении выравниваний аминокислотных или нуклеотидных последовательностей. Данный алгаритм реализуется программой пакета EMBOSS - needle. Она делает глобальное выравнивание последовательностей. Параметры настраиваемые в порграмме:
datafile - матрица весов аминокислотных или нуклеотидных замен, по умолчанию BLOSUM62 или DNAfull соответственно; gapopen - штраф за открытие гэпа (вес пропуска, который следует за аминокислотой), по умолчанию равен 10; gapextend - штраф за продолжение гэпа (вес пропуска, который следует за другим пропуском), по умолчанию равен 0,5; endopen - штраф за открытие гэпа (вес пропуска, который следует за аминокислотой), по умолчанию равен 10; endextend - штраф за продолжение гэпа (вес пропуска, который следует за другим пропуском), по умолчанию равен 0,5.
Программа water реализует алгоритм Смита-Ватермана, для локального парного выравнивания. В отличии от needle, программа находит оптимальное выравнивание участка последовательности, а остальную часть(которую needle заполняет гепами) убирает и не учитывает. В ходе работы water мы получаем только участок локального выравнивания с наибольшей массой.
Пример того как это работает:
При помоще скрипта evolve_protein.pl было сделано 3 мутанта (параметры: "-i" - указывает с последовательностью белка в fasta-формате, с которым мы собираемся работать; "-o" - файл, в который направятся результаты работы скрипта.
Мутанты удовлетворяют условия заданных в таблице:
Таблица 2. Критерии мутантов:
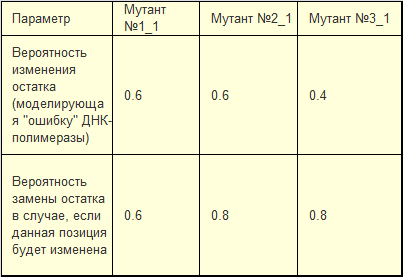
Таблица 3. Выравнивание для Мутанта№1_1.
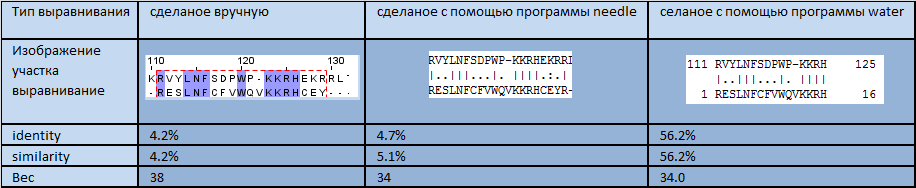
Таблица 4. Выравнивание для Мутанта№2_2.

Таблица 5. Выравнивание для Мутанта№3_3.
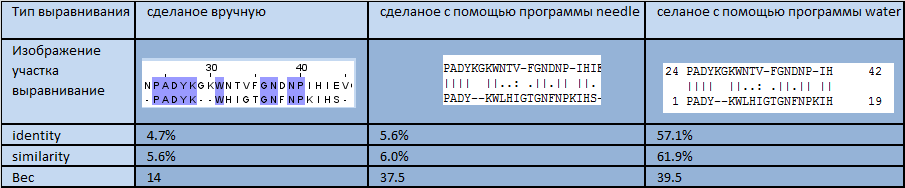
По результатам трех выравнивай выравниваний видно, что белок имеет длиную структуру и его локальные выравнивания более "весомые" и имеют лучше совпадение. Таблица 5 хорошо это подтверждает в случаи 50% identity у выравнивания сделаного water и ручного.
© Medvedev Dima 2012; дата последнего обновления 20.04.2013