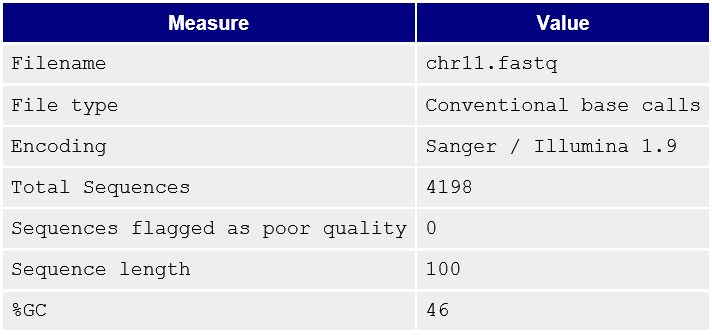
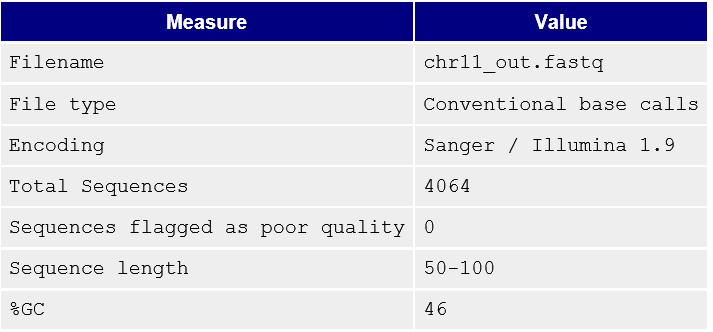
В данном задании анализировались риды из хромосомы 11 для чего использовалась хромосома 11 человеческого генома (сборка версии hg19).
В начале был сделан контроль качества ридов программой FastQC. В результате работы которрой был получен архив (.zip) и отчет о результатах работы программы в виде html страницы
команда: fastqc chr11.fastq
Далее была сделана очиста ридов с помощью программы Timmomatic. Были оставлены риды длиной не менее 50 нуклеотидов, и с конца каждого рида были удалены нуклеотиды с качеством ниже 20.
команда: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11.fastq chr11_out.fastq TRAILING:20 MINLEN:50
Для очищенного файла chr11_out.fastq опять был проведен контроль программой FastQC. Полученный отчет на html странице chr11_out_fastqc.html
В результате чистки из 4198 ридов осталось 4064.
Basic Statistics
| До чистки | После чистки |
| |
Per base sequence quality
| До чистки | После чистки |
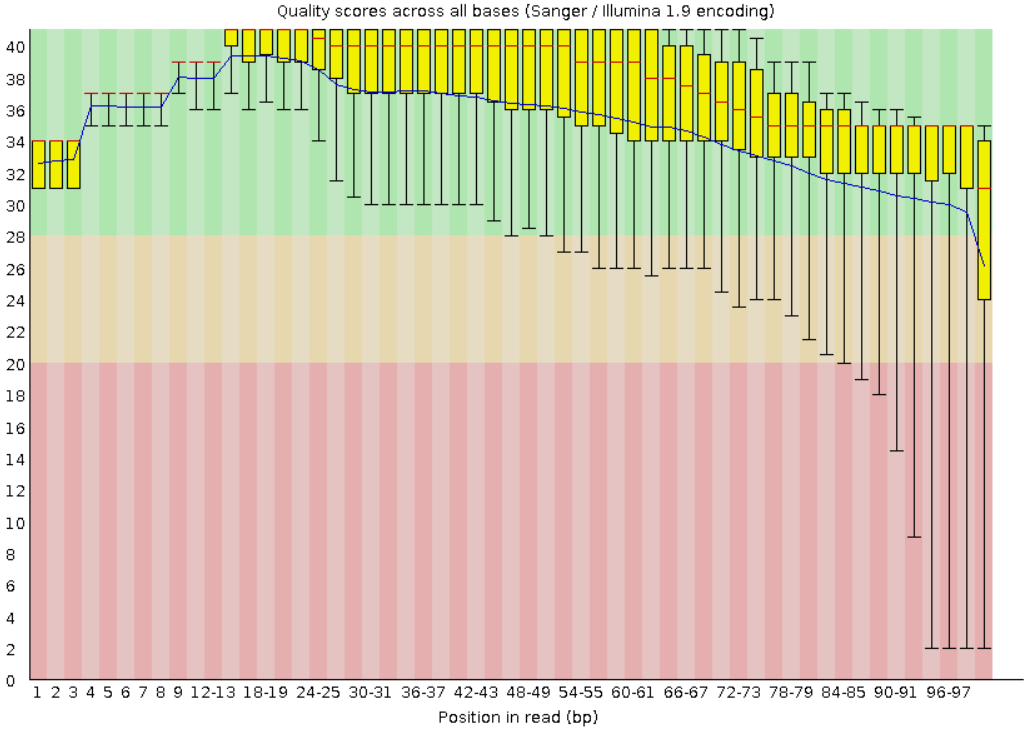 | 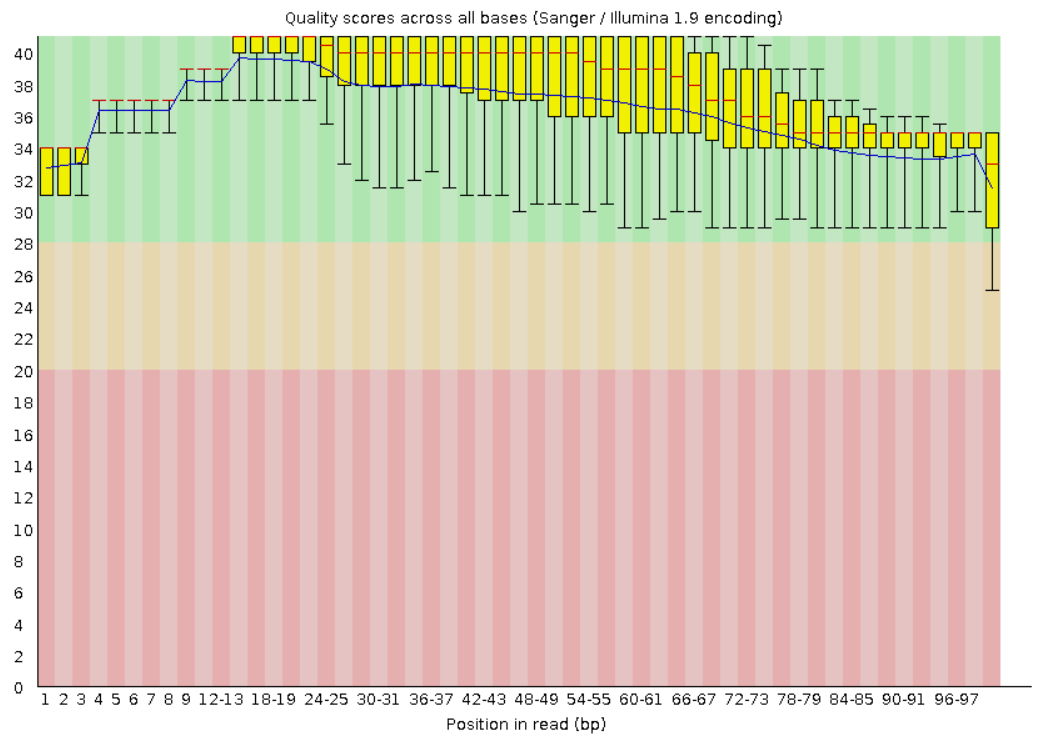 |
Видно, что после ридов качество чтений улучшилось. Были удалены плохие риды, а остались те, с которыми можно дальше работать.
Из других параметров практически все в порядке. Только 3 параметра до отчистки вызывают сомнения: Per base sequence content (содержание каждого нуклеотида в ридах), Per sequence GC content (содержание GC в ридах), Overrepresented sequences. И к ним еще один в случае очищенных ридов: Sequence Length Distribution (распределение длин ридов), что не удивительно, потому что мы же удалили риды длиной меньше 50 нуклеотидов.
Далее надо было картировать чтения на имеющуюся референсную хромосому.
Была проиндексирована референсная последовательность
команда: bwa index chr11.fasta
Было построено выравнивание прочтений и референса в формате .sam
команда: bwa mem chr11.fasta chr11_out.fastq > vyravniv.sam
Полученный в результате файл: vyravniv.sam
Выравнивание чтений с референсом было переведено в бинарный формат .bam.
команда: samtools view vyravniv.sam -b -o vyravniv.bam (-o - выходной файл; -b - бинарный формат)
Полученный в результате файл: vyravniv.bam
Выравнивание чтений с референсом (получившийся после картирования .bam файл) были отсортированы по координате в референсе начала чтения
команда: samtools sort vyravniv.bam -T vremyanka.txt -o sort.bam (-T - создает временный файл; -o - выходной файл)
Полученный в результате файл sort.bam
Отсортированный .bam файл был проиндексирован
команда: samtools index sort.bam
Было выяснено, сколько чтений откартировалось на геном
команда: samtools idxstats sort.bam > mapping.out
Полученный в результате файл: mapping.out
На хромосому из 4064 ридов откартировались все кроме 2ух.
Был создан файл с полиморфизмами
команда: samtools mpileup -uf chr11.fasta sort.bam > snp.bcf
Был создан файл со списком отличий между референсом и чтениями
команда: bcftools call -cv snp.bcf > snp.vcf
В этом файле записаны 23 отличия между референсной последовательностью и ридом. В основном, они представлены заменами (21) и только одной вставкой и одной делецией. Ниже представлена таблица с характеристиками трех полиморфизмов из этого файла.
| Полиморфизм | Координата | Тип | Референс | Чтение | Глубина покрытия | Качество чтения |
| №1 | 116650454 | Индель (делеция) | ATCTCT | ATCT | 196.468 | 22 |
| №2 | 116657590 | Индель (вставка) | СAAA | CAAAA | 144.467 | 63 |
| №3 | 17408630 | Замена | С | Т | 221.999 | 35 |
Надо было проанотировать с помощью программы annovar полученные SNP (без инделей) по базам данных refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Файл без инделей был переведен в формат avinput, с которым может работать annovar, с помощью скрипта convert2annovar.pl
команда: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_nonindel.vcf -outfile snp.avinput
Далее аннтоции были получены с помощью скрипта annotate_variation.pl
Refgene
Refgene - база данных для метода аннотации полиморфизмов, основанного на изменении строении генов и межгенных участков при инделях или заменах. Программа выдает 3 файла: в первом (variant_function) суммарная информация о всех найденных SNP, во втором (exonic_variant_function) содержится информация о SNP в экзонах, а в третем (log) информация о процессе работы программы.
команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr11_refgene -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
SNP были разделены на группы по положению в геноме:
При том гетерозиготных SNP 13, а гомозиготных 8
Имеющиеся 5 экзонных полиморфизмов расположены внутри 3ех генов:
Ген KCNG11. 2 несинонимичные замены (G748A:p.V250I; A67G:p.K23E). Ген кодирует интегральный мембранный белок входящего АТФ-зависимого калиевого канала. Мутации вызывают гиперинсулинная гипогликемия и инсулин независимый диабет.
Ген BUD13. 1 синонимичная замена (G480A:p.P160P) и 1 несинонимичная (C443T:p.P148L). Ген кодирует белок нужный для выведения мРНК из ядра и сплайсировании pre РНК.
Ген ZPR1. 1 несинонимичная замена (C791T:p.A264V). Ген кодирует белок с цинковым пальцем, учавствующий в дифференциации нейронов. Мутации вызывают апоптоз моторных нейронов и спинальную мышечную атрофию.
dbsnp
dbsnp - база данных уже аннотированных полиморфизмов. Командва выдает 3 файла: первый (dropped) с SNP с идентификаторами rs, второй (filtered) с SNP без rs. и третий (log) с информацией о работе программы.
команда:perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Получилось, что 19 из 21 SNP аннотированы в dbsnp
1000 genomes
1000genomes - база данных полиморфизмов из 1000 геномов людей. Команда выдает 3 файла аналогичные предыдущему пункту, только для базы 1000genomes.
команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11_1000genomes -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Получилось, что в данной базе не аннотированы те же 2 SNP, что и в предыдущей.
GWAS
GWAS - база данных аннотированных полиморфизмов, которые ассоциированы с заболеваниями или особыми последствиями. Команда выдает 2 файла: первый (gwasCatalog ) с SNP, с известными клиническими значениями, и второй (log) c информацией о работе программы.
команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr11_gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
У 6 SNP были определены как клинически значимые. 2 связаны с диабетом 2ого типа; 2 связаны с увеличенным количеством триглицеридов (препятсвуют кровотоку); 1 связан с метаболическим синдромом; 1 связан с ожирением.
Clinvar
Clinvar - база данных о о связи между полиморфизмом и его влиянием на организм человека (патогенный, неизвестный, отвечающий на лечение лекарствами и др.)
Команда выдает 3 файлa: первый (dropped) с аннотировнными SNP, второй (filtered) с остальными SNP и третий (log) с информацией о работе программы.команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11_clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
2 SNP аннотированы в этой базе данных. 1ый как непатогенный, неспецифицированный и 2ой как непатогенный, отвечающий за лечение диабета 2ого типа.
Общая таблица с найденными SNP table