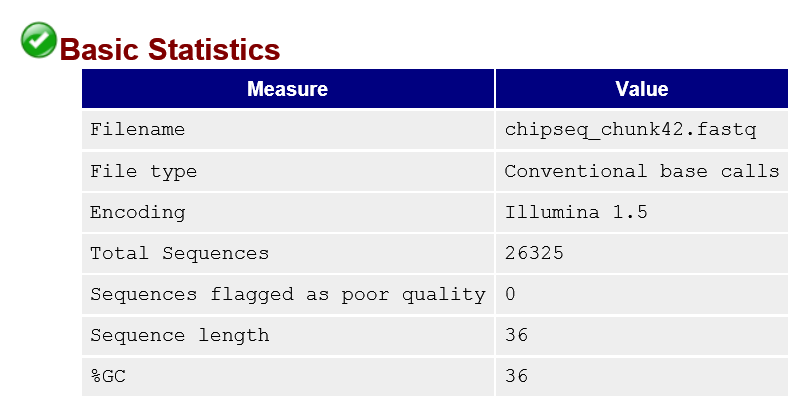
Для данного практикума были взяты риды, полученные в результате chip-seq эксперимента из файла chipseq_chunk42.fastq
Для оценки качества ридов использовалась программа FastQC
| fastqc chipseq_chunk42.fastq |
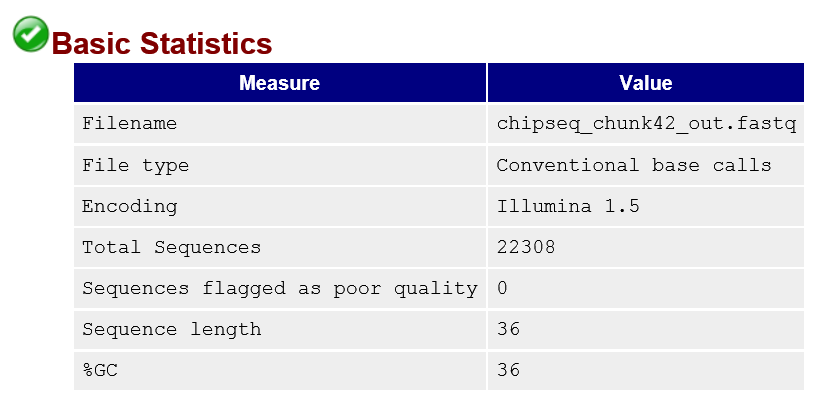
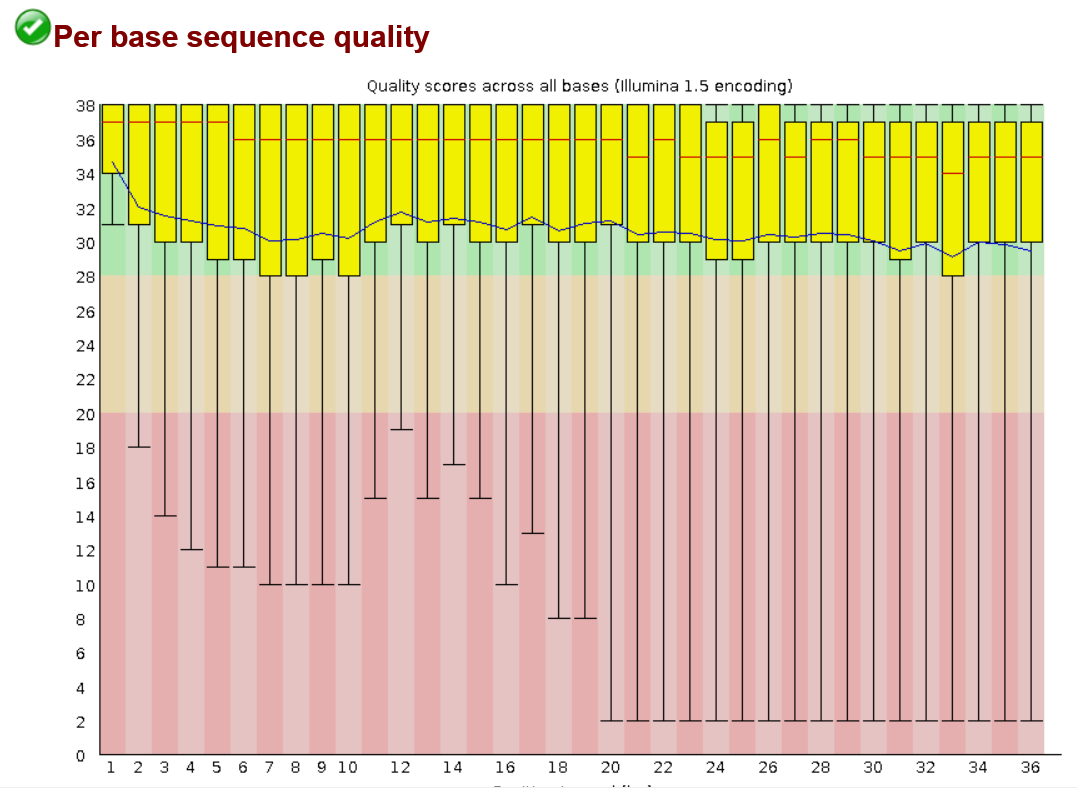
Полученный html файл По результатам работы этой программы, несмотря на то, что ридов с плохим качеством нет, было принято решение удалить все риды с длиной менее 36 п.н. и качеством ниже 50 с помощью программы Trimmomatic
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk42.fastq chipseq_chunk42_out.fastq TRAILING:50 MINLEN:36 |
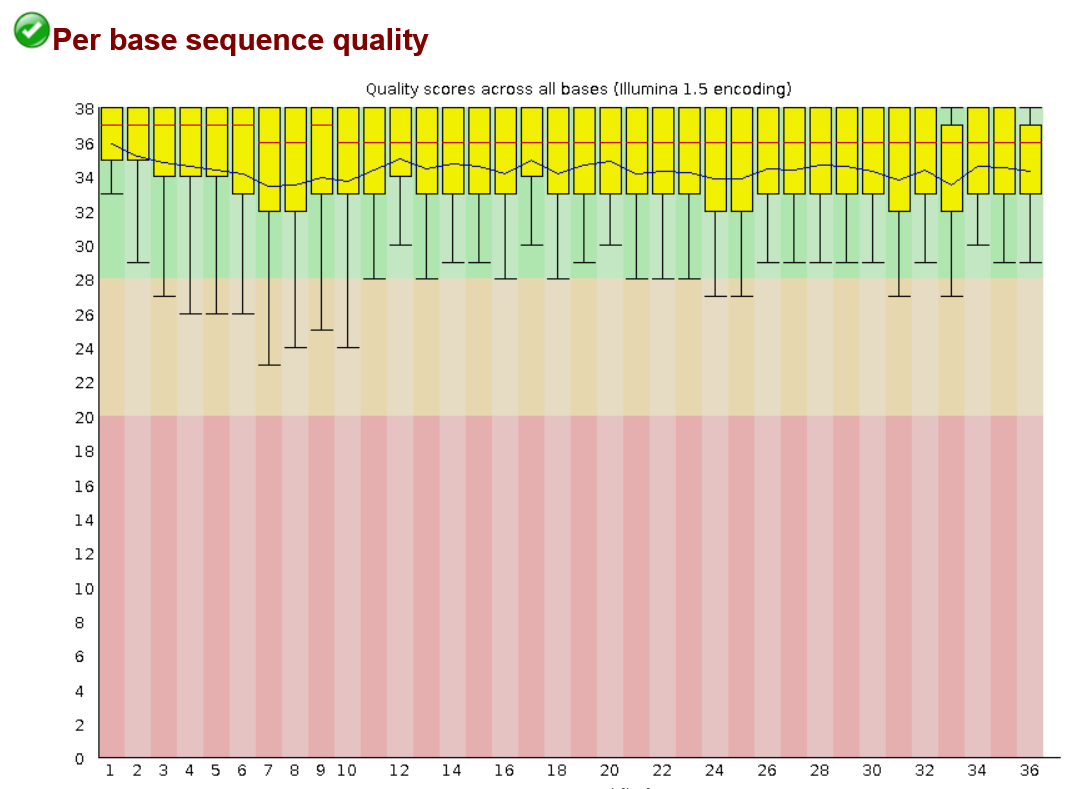
Отчищенный риды опять были проанализированы с помощью программы FastQC. Полученный html файл Всего было удалено 15,3% (4017) ридов
| |
| |
| Рис1. Риды до чистки | Рис2. Риды после чистки |
Картирование проводилось программой BWA для референсного генома (уже проиндексированного).
| bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk42_out.fastq > chipseq_chunk42.sam |
Потом с помощью пакета Samtools файл с выравниванием был переведен в бинарный формат
| samtools view chipseq_chunk42.sam -bSo chipseq_chunk42.bam |
Далее полученное выравнивание ридов было отсортировано по координатам начала ридов на референсе. Параметр -Т организует запись временный данных, чтобы они не ввыводились в stdout.
| samtools sort chipseq_chunk42.bam -T chip_temp -o chipseq_chunk42_sorted.bam |
Полученный файл был проиндексирован
| samtools index chipseq_chunk42_sorted.bam |
Далее было выяснено, сколько ридов откартировалось на геном. Полученный файл записан в chipseq.out
| samtools idxstats chipseq_chunk42_sorted.bam > chipseq.out |
Из 22308 ридов на геном откартировалось 22286 ридов - 99,9%, при том 21727 ридов (97,5% от откартированных ридов) откартировались на 20 хромосому. Таким образом можно утверждать, что мне была дана 20 хромосома.
Искались участки генома, на которые откартировалось наибольшее число ридов. В случае эксперимента chip-seq эти участки будут соответствовать участку сайта связывания ТФ, который был защищен этим ТФ.
Была использована команда MACS (Model-based Analysis of Chip-Seq), т.к. число пиков было слишком маленьким, то с параметром --nomodel
| macs2 callpeak -n chipseq_chunk42 -t chipseq_chunk42_sorted.bam --nomodel |
В результате работы этой программы были получены 3 файла. chipseq_chunk42_summits.bed chipseq_chunk42_peaks.narrowPeak chipseq_chunk42_peaks.xls Было найдено 13 пиков на 20 хромосоме (см. Таблицу)
| № | Начало | Конец | Длина | S между началом и вершиной пика | -lg(p-value) |
| 1 | 7565763 | 7566048 | 286 | 164 | 28.76848 |
| 2 | 8075333 | 8075563 | 231 | 192 | 8.84892 |
| 3 | 8125310 | 8125688 | 379 | 173 | 31.87013 |
| 4 | 8136053 | 8136277 | 225 | 168 | 14.64740 |
| 5 | 8137179 | 8137666 | 488 | 207 | 35.69110 |
| 6 | 8186666 | 8186899 | 234 | 153 | 18.21723 |
| 7 | 8231191 | 8231663 | 473 | 181 | 71.04403 |
| 8 | 8311608 | 8312006 | 399 | 142 | 36.79707 |
| 9 | 8395664 | 8395992 | 329 | 183 | 23.88743 |
| 10 | 8445126 | 8445330 | 205 | 150 | 16.89913 |
| 11 | 8861157 | 8861422 | 266 | 100 | 16.63130 |
| 12 | 9266370 | 9266569 | 200 | 44 | 12.10036 |
| 13 | 9282520 | 9282766 | 247 | 87 | 31.43082 |
Визуализация пиков проводилась в UCSC Genome Browser Ему был подан файл chipseq_chunk42_peaks.narrowPeak, в котором в начале было написано: " track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 42" browser position chr20:7565762-928276 "
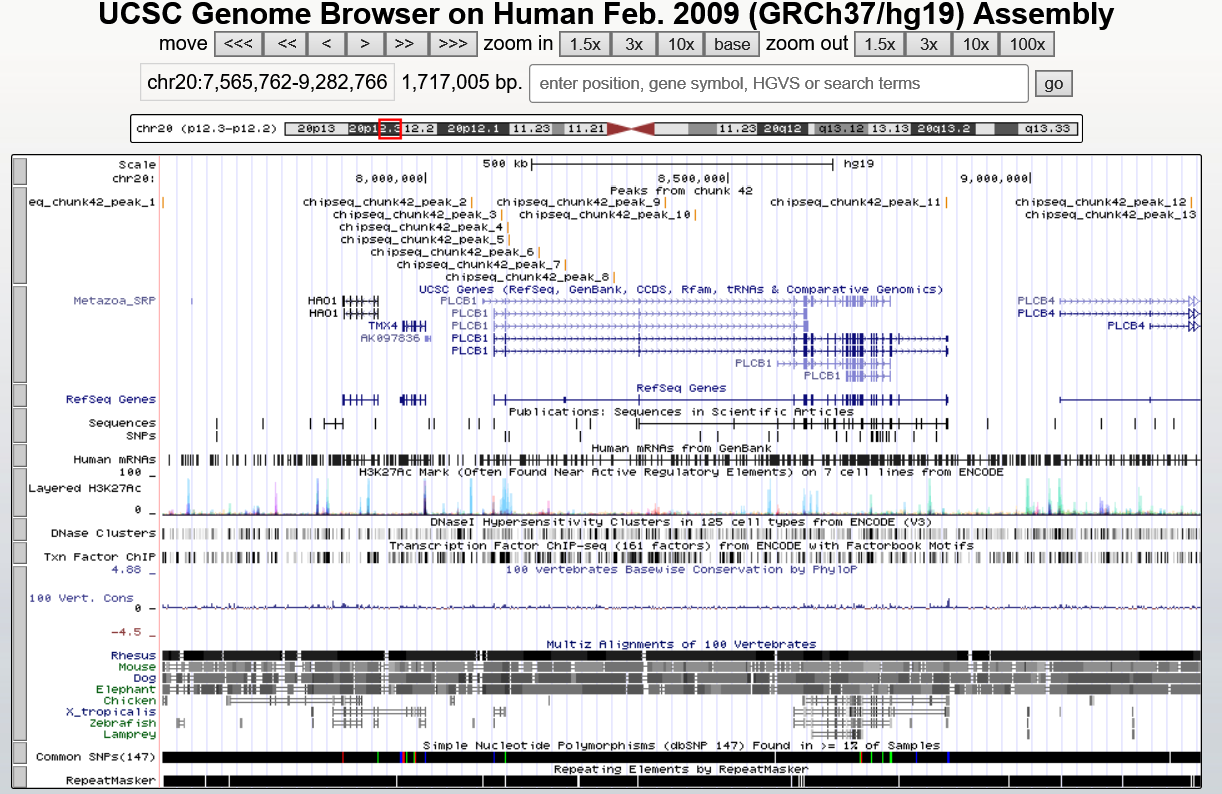
Рис3. Визуализация пиков в UCSC Genome Browser
Среди имеющихся пиков только один (№7) имеет значительно больший -lg(p-value), т.е. значительно меньший p-value, а значит этот пик наиболее достоверен. Этот пик и визуализирована на Рис4.
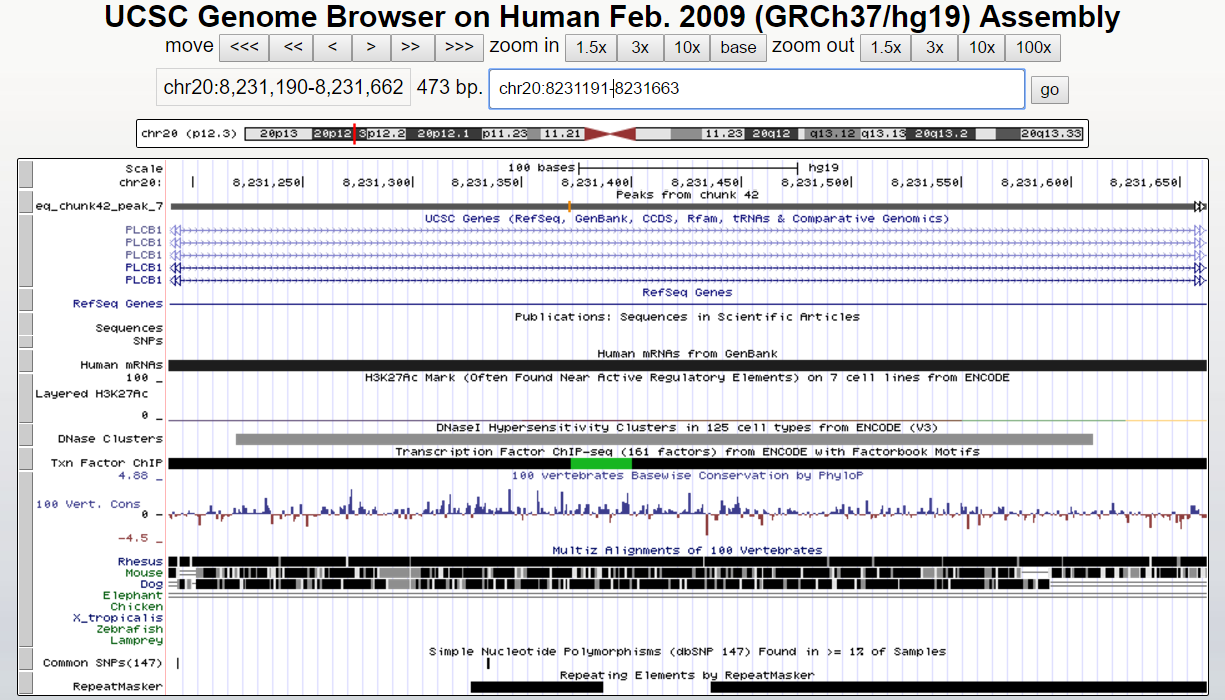
Рис4. Наиболее достоверный пик
Как мы можем видеть, пик картировался на интрон гена PLCB1: phospholipase C beta 1. Белок, кодируемый этим геном, катализирует формирование инозито 1,4,5-трифосфата и диацилглицирола из фосфатидилинозитола 4,5-бифосфата. Кофактором реакции является кальций. Эта реакция важна для передачи внтуриклеточных и межклеточных сигналов. Ген активируется двумя альфа-субьединицами alpha-q и alpha-11 G-белка.