

Для выполнения данного практикума был выбран домен Acetyltransferase_13 : ID - Acetyltransf_13; AC - PF13880. Ацетилтрансферазы переносят ацетильную группу на молекулу субстрата. В этом домене 16 разных архитектур, представленных 750 последовательностями у 594 видов. Известно 3 3D структуры.
Ссылка на страницу домена Pfam
Ссылка на доменные архитектуры
Для данного домена было посторено выравнивание с помощью JalView: File → Fetch Sequences → Pfam (Full) → PF13880. Выравнивание было покрашено ClustalX, by conservation 30%. В выравнивании можно выделить два относительно консервативных блока, разделенных участом гепов. Также к выравниванию было добавлено изображение 3D струкуры белка ESCO1_HUMAN (PDB ID 4MXE).
Для анализа были выбраны две доменные архитектуры. Первая с двудоменная: zf-C2H2_3, Acetyltransf_13, представленная 655 последовательностями (рис1) и вторая однодоменная: Acetyltransf_13, представленная 72 последовательностями (рис2)
| |
| Рис1. Архитектура1. Зеленым - zinc-finger, красным - Acetyl-transferase | Рис2. Архитекура2. Зеленым - Acetyl-transferase |
С помощью скрипта swisspfam_to_xls.py из файла с информацией об архитектуре всех последовательностей Uniprot /srv/databases/pfam/swisspfam.gz была извлечена данная информация про исследуемый домен.
| python swisspfam_to_xls.py -z -i /srv/databases/pfam/swisspfam.gz -p PF13880 -o domens.xls |
Далее была составлена сводная таблица: по строкам АС последовательностей, по столбцам домены.
Из Uniprot были скачены все последовательности (Uniprot → Retrieve, сохранен flat text). Из полученного файла с помощью скрипта uniprot_to_taxonomy.py была получена таксономия.
| python uniprot-to-taxonomy.py -i uniprot.txt -o taxonomy.xls |
Полученная таксономия была добавлена в имеющуюся Excel таблицу с помощью функции ВПР. Так же в эту таблицу была добавлена длина последовательности.
В качества таксона были выбраны Eucaryota, а как подтаксоны Metazoa и Fungi (в названиях они отражаются буквами M и F). И для каждой архитектуры были выбраны по 10-12 последовательностей из каждого таксона, при том длины выбранных последовательностей специально брались примерно одинаковые (в районе 70), для того, чтобы выравнивание можно было бы сделать лучше. Они отмечены "+"
Итоговая таблица по архитектурам
Далее с помощью скрипта были получены два файла: 1 c идентификатарами выбранных последовательностей, 2 с теми же идентификаторами, только с указанием архитектуры и таксона. По идентификаторам 1ого файла с помощью скрипта filter-alignment.py было отредактированно полученное ранее выравнивание, так чтобы там остались только выбранные последовательности.
| python filter-alignment.py -i vyrav.fa -m ID_selected.txt -o vyrav_select.fa -a "_" |
Потом с помощью еще одного скрипта в названия последовательностей в выравнивании была добавлена информация об архитектуре (цифра 1 или 2) и таксоне (буква М или F).
С этим выравниванием и проводилась работа. Были удалены все пустые колонки, частично N, C концы. Какие-то целые последовательности было решено не удалять, т.к. сильно выбивающихся и портящих картину не было. Группа с первой архитектурой была покрашена ClustalX, by conservation 30%, а со второй архитектурой BLOSUM62 by concervation 30%. (рис3.)
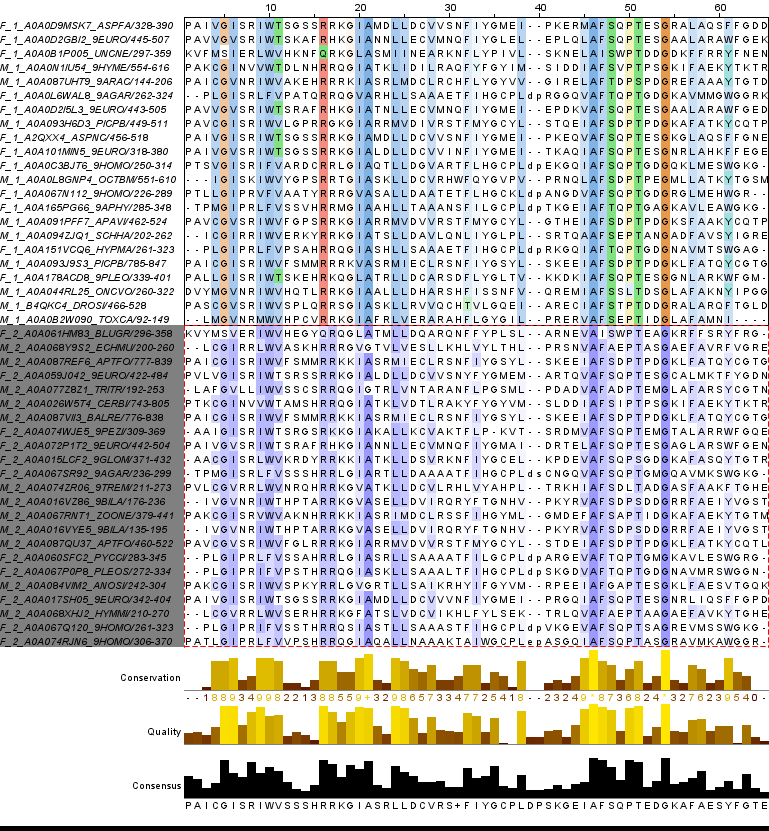
Рис3. Итоговое выравнивание
Как можно видеть и обеих групп по краям (но не по самым) существуют два относительно конесервативных блока. А по центру между ними довольно произвольная последовательность. Можно отметить одну абсолютно консервативную колонку из "G", одну где среди всех "G" по одному разу в обеих архитекстурах встречается "S", одну, где среди всех "F" в обеих архитектурах по одному раз встречается "I". В целом вторая архитектура чуть-чуть менее консервативная: в двух абслютно конесеравативных для архитектуры1 колонках с "А" и одной с "Т" во второй архитектуре совсем немного, но встречаются другие аминокислоты. Но в общем выравнивание можно считать достаточно достоверным, чтобы сроить по нему дерево.
Построение филогенетического дерева проводилось программой МЕГА при том двумя алгоритмами Neighbour-joining (рис4) и Maximum likelihood (рис5) со 100 bootstrap репликами.
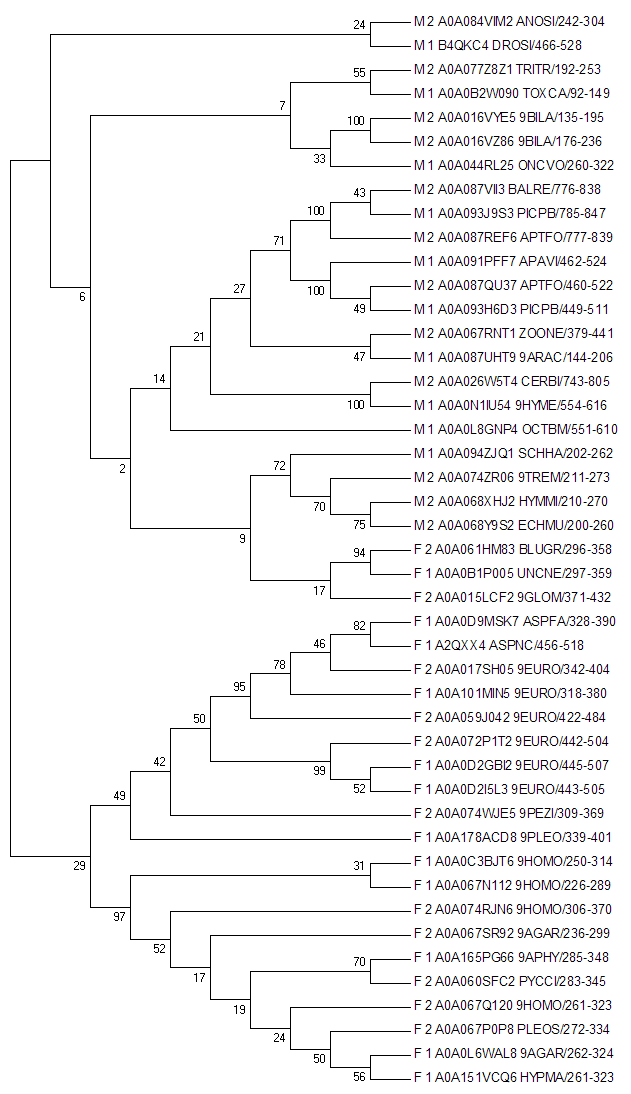 |  |
| Рис4. Neighbour-joining | Рис5. Maximum likelihood |
Данное дерево довольно неплохо разделило таксоны, по этой причине оно было укоренено в ветвь, разделяющую Metazoa и Fungi. Разделение на два подтаксона происходит полностью, за исключением трех последовательностей грибов, которые оказались одинаковыми для обоих методов. При том внутри таксонов какого-то четкого разделения на архитектуры не наблюдается. Т.е. нельзя сказать, что происходили какие конкретные значимые эволюционные события.