Практикум 15. De novo сборка генома
Основное задание
Задачей данного практикума является de novo сборка генома бактерии Buchnera aphidicola из чтений, имеющих ID: SRR4240361.
Чтения были скачаны из базы EMBL коммандой :
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/001/SRR4240361/SRR4240361.fastq.gz
Прежде чем начать сборку, чтения необходимо триммировать - для этого был использован trimmomatic с опицей -SE (опция для работы с одноконцевыми чтениями) .Сначала я избавился от адаптеров командной :
java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 15 SRR4240361.fastq.gz SRR4240361_no_adapters.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7
В результате из 7272621 ридов осталось 7238089 , т.е. было выброшено 0.47% ридов. Затем я удалил с правых концов чтений нуклеотиды с качеством ниже 20 и удалил все риды длинной ниже 32. Команда :
java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 15 SRR4240361_no_adapters.fastq.gz SRR4240361_good.fastq.gz TRAILING:20 MINLEN:32
В результате из 7238089 ридов осталось 6834335, т.е. было потеряно 5.58% ридов.
Далее из тримированных чтений были получены k-меры длинной 31, которые были использованы для сборки генома. Команды :
velveth ./SRR4240361_velvet 31 -fastq.gz -short SRR4240361_good.fastq.gz
velvetg ./SRR4240361_velvet
В результате сборки было получено 474 контигов. N50 сборки составил 25713 bp (то, что программа выдает длины в количестве k-меров учтено). Аномальных покрытий не наблюдается. Самые длинные контиги представлены в Таб.1 .
| Контиг | Длина контига | Покрытие |
| NODE_6_length_49238_cov_26.660851 | 49238 | 26.66 |
| NODE_2_length_45555_cov_26.450466 | 45555 | 26.45 |
| NODE_2_length_43866_cov_23.514977 | 45555 | 23.514977 |
Затем последовательности этих контигов были выравнены на геном нашего организма с помощью megablast (wordsize= 28) и blastn (wordzise=7). ID генома: CP009253
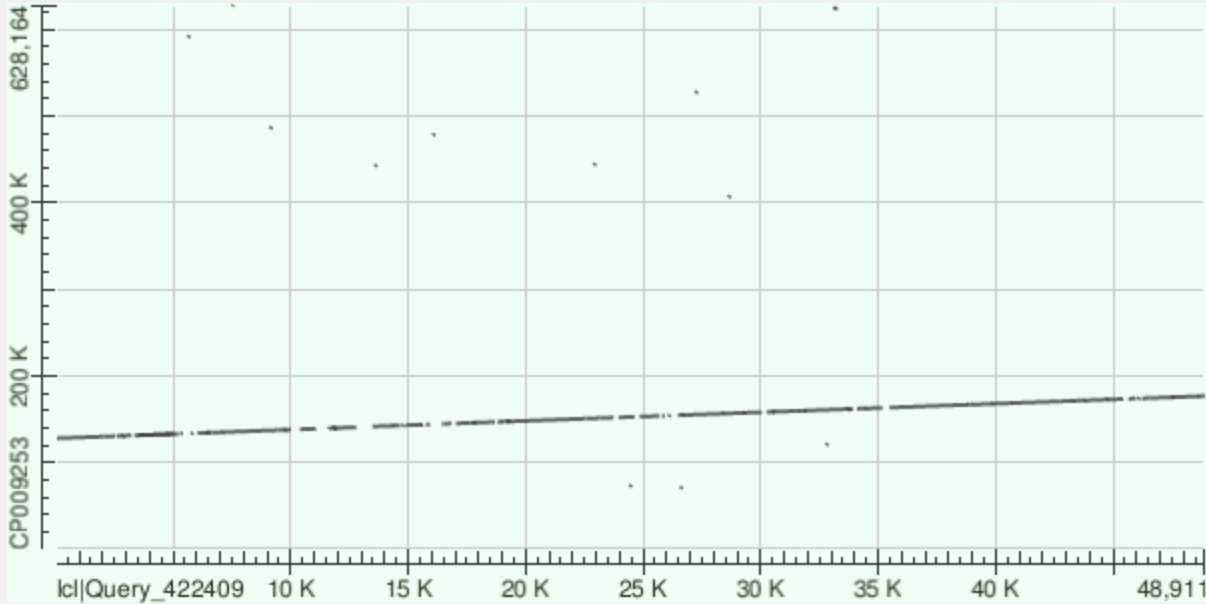
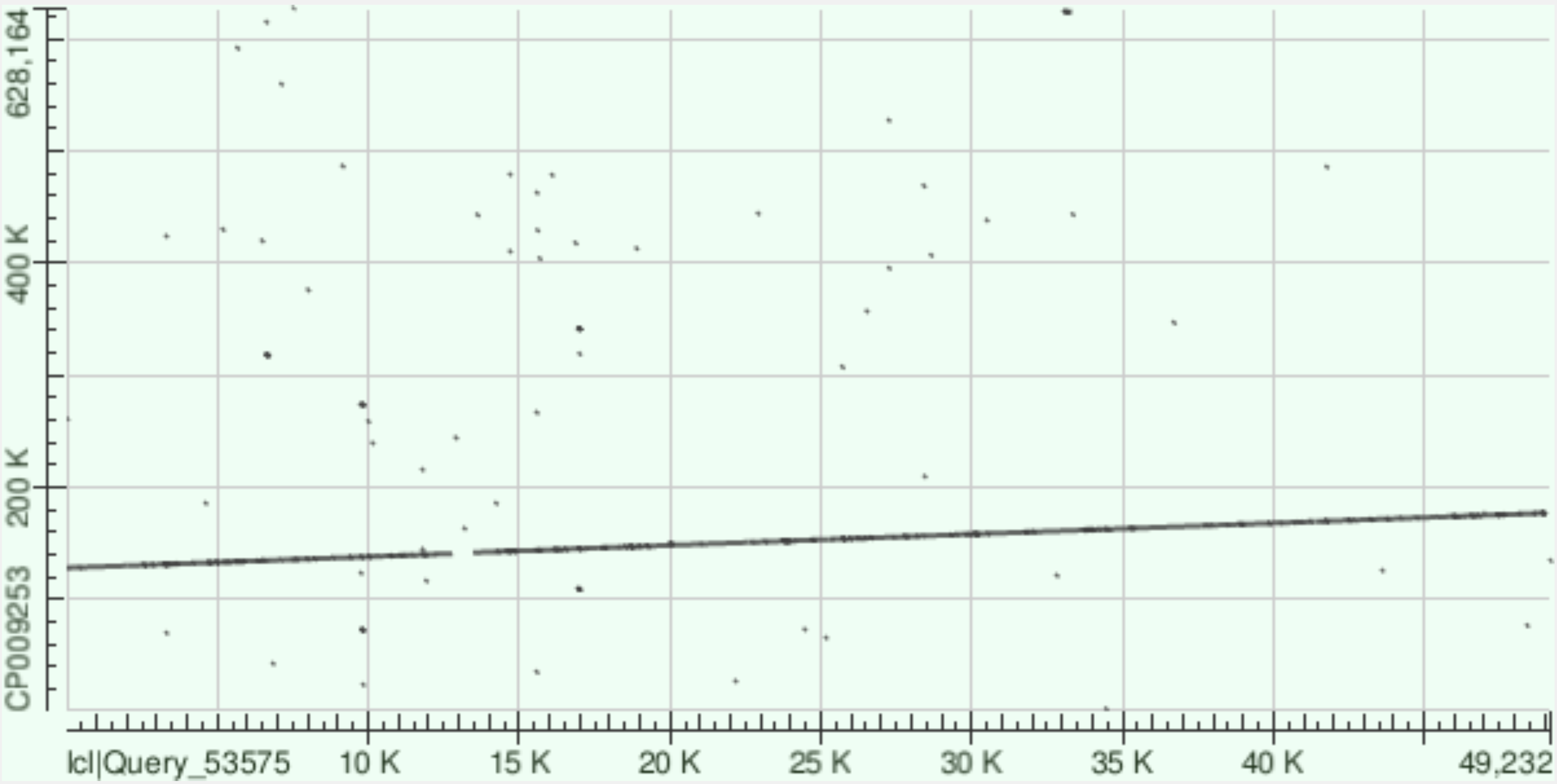
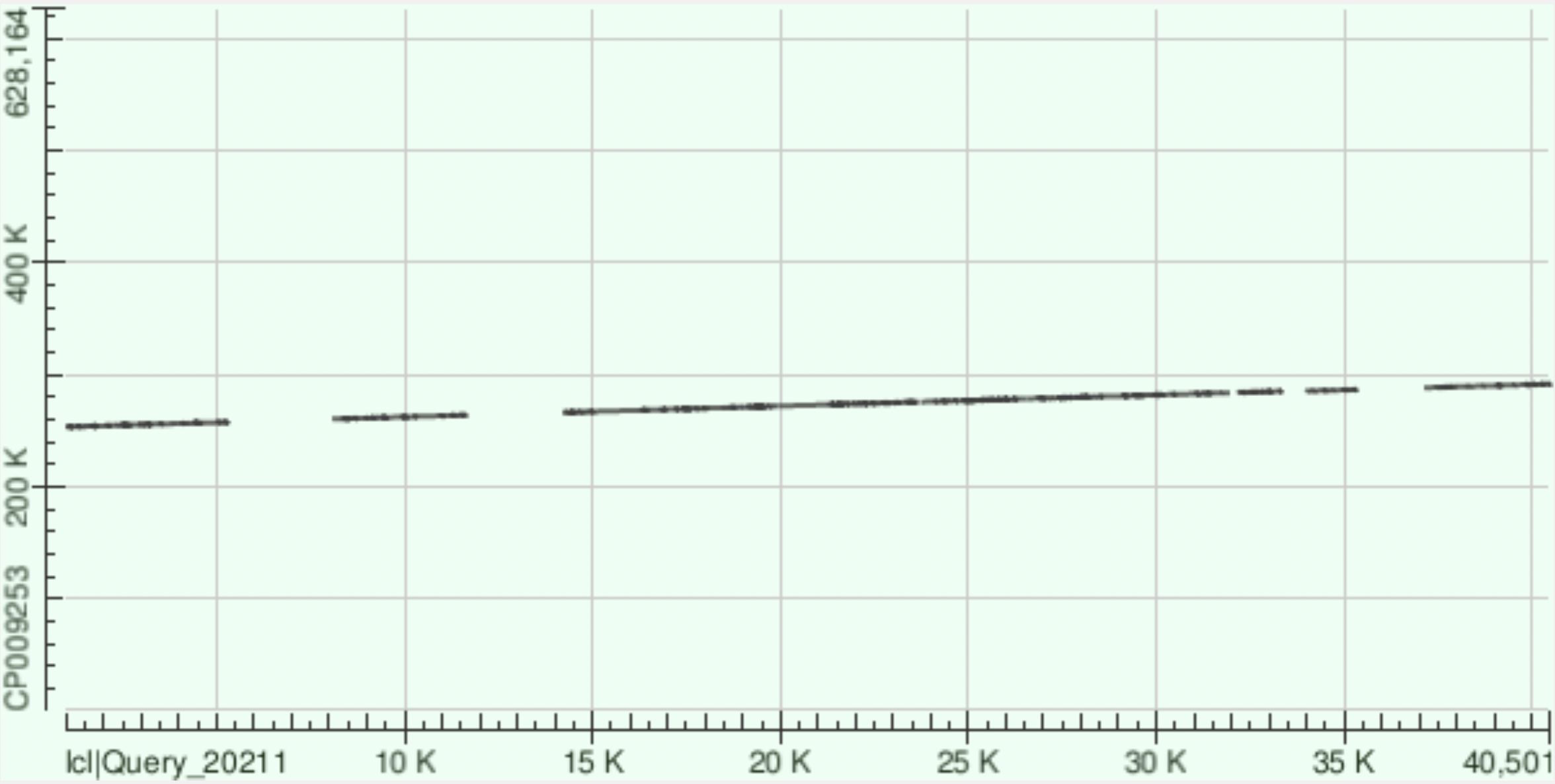
На Рис1 и Рис2 показаны Dot Plot выравнивания NODE_6_length_49238_cov_26.660851 с геномом алгоритмами megablast и blastn. На dot plot полученном megablast последовательности довольно хорошо выравнены. Имеется лишь немного пустых участков. Но особо крупных инверсий и делеций не наблюдается. Если посмотреть на карту, полученную blastn, то некоторые из этих пустых участков выравнивались, что говорит о том, что там есть мисмэтчи.
Информацию о выравнивании контига можно увидеть в Hit Table файле, так как у меня получилось многовато(45) выравниваний и делать из них таблицу затруднительно.
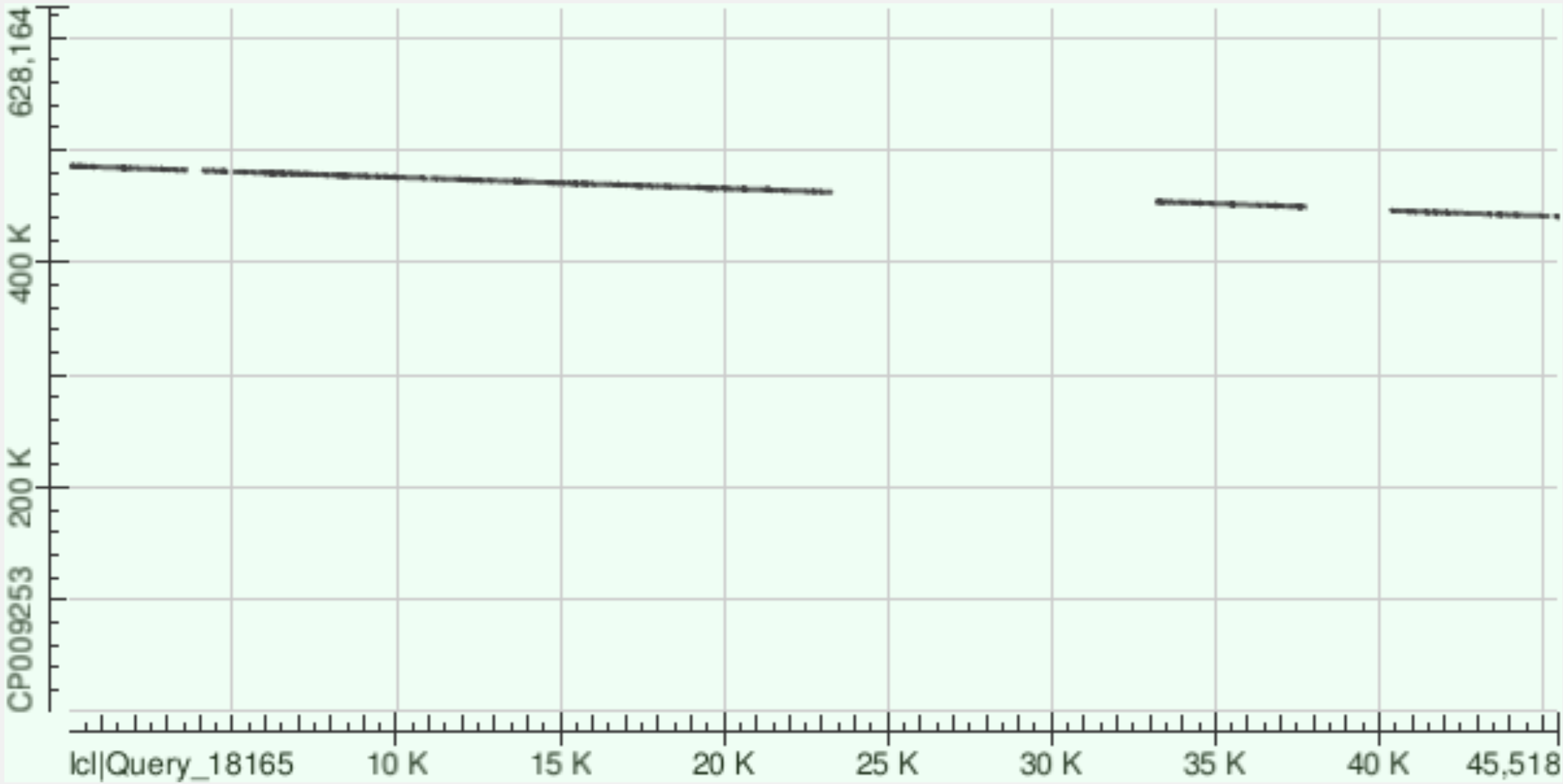
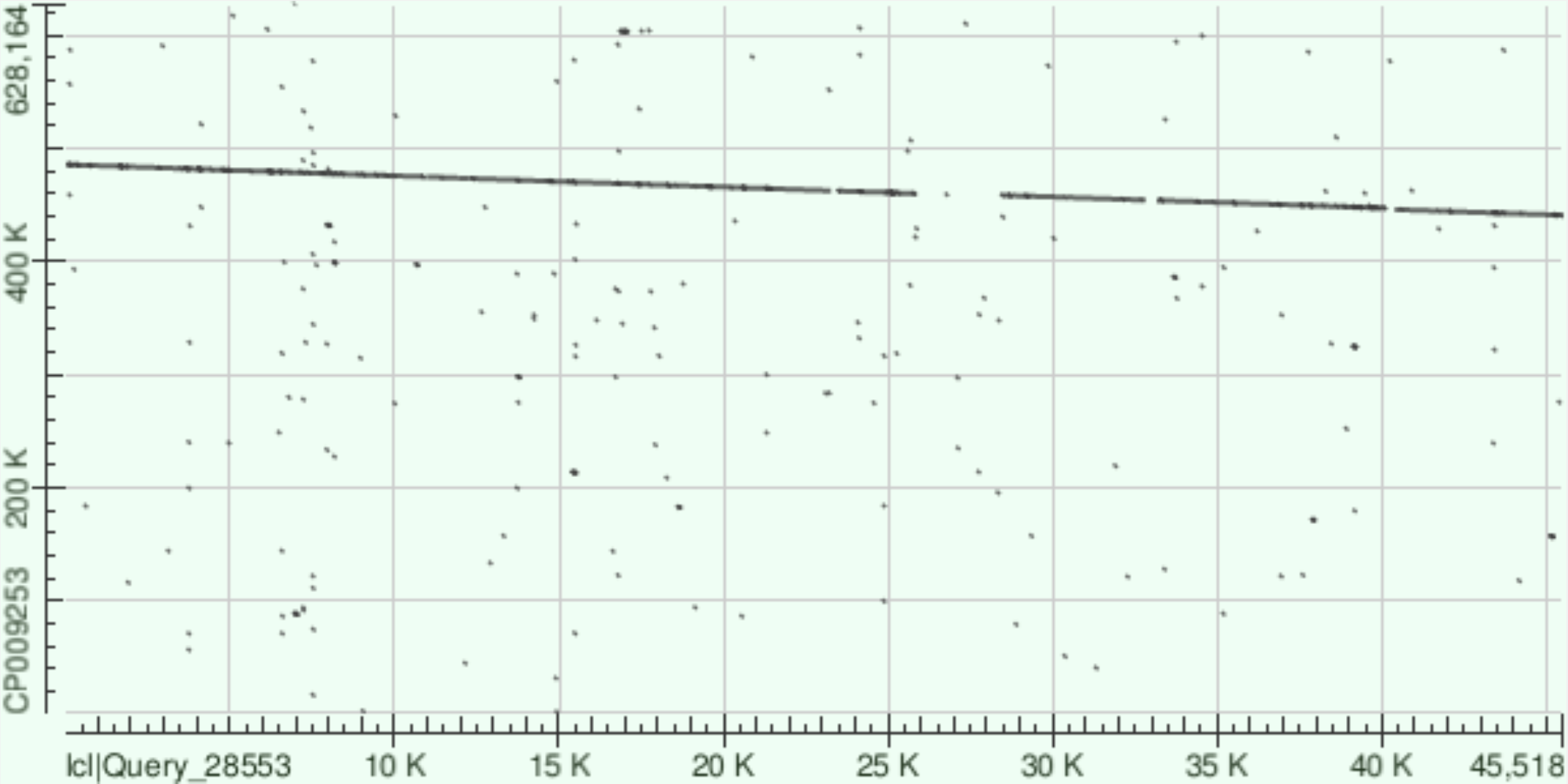
Этот контиг тоже довольно хорошо выравнился. Аналогично предыдущей карте локальных сходств имеюся пустые участки, которые выравниваются в blastn. Здесь линия выравнивания идет сверху вниз, что говорит о выравнивания с комплементарной цепью. Либо же этот участок подвергся инверсии. Учитывая длину контига такой вариант вполне возможен. Если смотреть на выдачу blastn, то есть участок между 26k-28k по оси абсцисс, который не выравнялся. Пристально посмотрев у меня появилось ощущение, что там есть сдвиг графика, т.е произошла делеция. Если посмотреть на Hit Table данные о выравнивании и на карту локального сходства, то можно увидеть, что выравнивание заканчивается на координатах [25826,460011], а продолжается с [28447,458588]. 28447-25826=2621. Получается относительно оси ординат график должен упасть на 2621, прежде чем заново начать выравниваться.Но у меня выходит |458588-460011|=|1423|.Получается, что случился сдвиг, которые, возможно, является делецией. (я надеюсь это так работает)
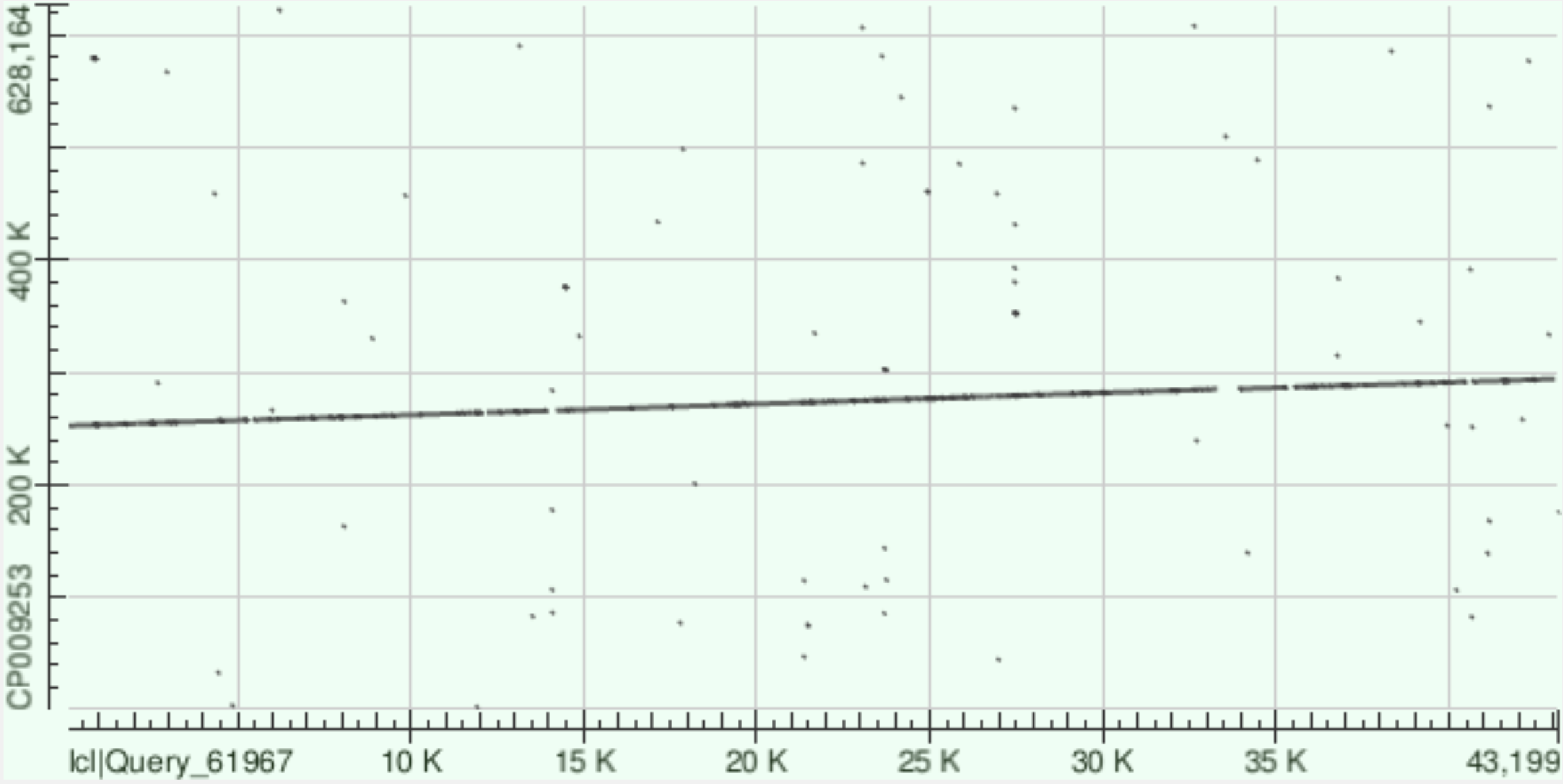
Файл с информацией о выравнивании. Контиг очень хорошо выравнился с участком генома, хоть и имеются некоторые пустые участки. Цепи были выбрано одинаково. При просмотре карты выданной blastn многие пустые участки выравнились, что говорит о наличии там мисмачтей.
Дополнительное заданин. k-меры 27
В рамках первого доп.задания я из тримированных чтений получил k-меры длинной 27 и использовал их для сборки. В результате сборки N50 оказалось ниже - 15564. Теперь самыми длинными контигами являются :
| Контиг | Длина контига | Покрытие |
| NODE_14_length_50427_cov_46.11 | 50427 | 46.114819 |
| NODE_4_length_39324_cov_41.74 | 39324 | 41.749313 |
| NODE_1_length_34494_cov_45.15 | 34494 | 45.153882 |
Можно заметить, что в сравнении с прошлой сборкой эти контиги короче (кроме самого длинного), а покрытие у них больше. В целом, контигов стало больше. Если при прошлой сборке их было 474, то сейчас 2531. Можно сделать вывод, что при понижении длины k-меров сборка стала хуже.
Дополнительное заданин. Другие параметры триммирования
В рамках данного задания я пробовал иначе триммировать свои чтений (уже избавленные от адаптеров). Я использовал параметр SLIDINGWINDOW:5:20, который проходит по каждому чтению окном длины 5 нуклеотидов, и если среднее качество какого-либо участка чтения оказывается ниже 20, то удаляются эти 5 букв и все что, правее. Команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 15 SRR4240361_no_adapters.fastq.gz SRR4240361_good.fastq.gz SLIDINGWINDOW:5:20
В результате запуска из 7238089 чтений осталось 7065728, т.е. было потеряно 2.38%.
Теперь из этих чтений я получаю k-меры длинной 31 и собираю геном. В результате сборки N50 получился 25713 (точно такой же, как и при первой сборке).В целом результаты сборки очень схожи с первой сборкой. Общее число образовавшихся контигов - 406. Двойка самых длинных контигов имеют точно такую жу длину, как и при первой сборке. Лишь немного изменилось их покрытие. (Таб.3)
| Контиг | Длина контига | Покрытие |
| NODE_6_length_49238_cov_26.203481 | 49238 | 26.20 |
| NODE_2_length_45555_cov_26.018329 | 45555 | 26.01 |
| NODE_32_length_43866_cov_23.069667 | 43866 | 23.06 |
В целом можно сделать, вывод что качество сборки не сильно изменилось при использовании другого пути триммирования чтений.
Дополнительное задание. Сборка программой SPAdes
В рамках этого задания для сборки генома я пользуюсь прораммой SPAdes. При этом для сборки я использую k-меры длинной 31 (как и в основном задании) и соответсвенно сравнивать я буду сборку из основного задания и ту, которую сделаю здесь. Команда для сборки:
spades.py -s SRR4240361_good.fastq.gz -k 31 --only-assembler -o SRR4240361_SPAdes
В таблице 4 показаны некоторые характеристики сборок, как число образовавшихся контигов, N50 (учтено, что теперь программа выдает длину в количестве нуклеотидов), длины самых длинных контигов и их покрытие. В результате сборки программой SPAdes получается значительно меньше контигов и более высокое значение N50, чем в результате сборкой программой velvet. Из этого я делаю вывод, что сборка полученная SPAdes превосходит сборку полученную velvet.
| Программа | N50 | Длина самого длинного контига | Покрытие самого длинного контига | Число контигов |
| Velvet | 25713 | 49238 | 26.66 | 474 |
| SPAdes | 30583 | 49269 | 21.87 | 122 |