| Главная |
| Семестры |
| О себе |
| Ссылки |
1. Выбор белка и структур
Для выполнения задания из списка белков, для которых есть структура, полученная методом РСА
и структура, полученная методом ЯМР, был выбран белок CheY. Белок CheY необходим бактериям
для хемотаксиса, и изучался рентгеноструктурно из-за больших конформационных изменений,
вызванных связыванием белка с ионом магния.
Рентгеновская структура белка (идентификатор 1CHN)
CheY имеет разрешение 1.76 Ангстрем и R-фактор 0.191. Таким образом,
это достаточно хорошая структура.
ЯМР-структура, полученная через 7 месяцев после рентгеноструктурной другой группой
исследователей, содержит 46 моделей. Идентификатор структуры: 1CEY.
Обе структуры выбранного белка показаны на рисунке 1.
A. 1CHN |
B. 1CEY |
 |
 |
Рисунок 1. Сртуктуры белка CheY, полученные методами РСА (А) и ЯМР (В)
Можно заметить, что структуры достаточно похожи, взаиморасположение всех структурных элементов (альфа-спиралей, бета-листов и петель) примерно одинаковое.
2. Выбор водородных связей
В двух выбранных структурах были рассмотрены три водородные связи в разных частях белка:
1. Связь в петле между азотом 88 аланина и кислородом 91 лизина.
2. Связь между аргинином в альфа-спирали и глутаматом в бета-листе.
3. Связь между 67 глутаматом и 71 треонином, которые образуют альфа-спираль.
Наличие водородной связи определялось по структуре, разрешенной РСА, и длина ее не должна была превышать
3.5 Ангстрем. Расположение выбранных аминокислот в структурах белков показано на рисунке 2.
A. 1CHN |
B. 1CEY |
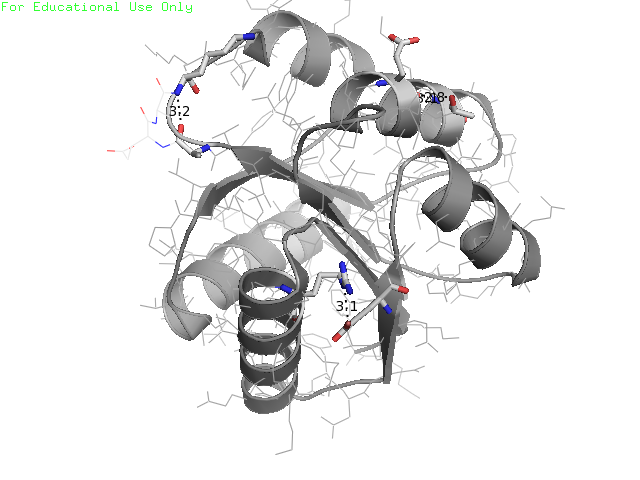 |
 |
Рисунок 2. Выбранные аминокислоты в структурах белка. На рисунке А выбранные аминокислоты обозначены более жирными палочками, а на рисунке В выделены красным и палочками.
3. Сравнение водородных связей
A. 1CHN A88-K91 |
B. 1CEY A88-K91 |
 |
 |
C. 1CHN R18-E35 |
D. 1CEY R18-E35 |
 |
 |
E. 1CHN E67-T71 |
F. 1CEY E67-T71 |
 |
 |
Рисунок 3. Выбранные водородные связи в структурах белка.
Структуры, полученные методом ЯМР, особенно ценны, так как отображают реальное положение всех аминокислот в растворе и дают представление о том, какие участки молекулы белка более подвижны, а какие -- менее. Можно заметить, что водородная связь, присутствующая во внешней петле в РСА-структуре, в растворе не существует, хотя положение самой петли относительно фиксировано -- нет сильно отстоящих петель в различных ЯМР-моделях. Водородная связь в белковой глобуле между α-спиралью и β-листом присутствует не во всех ЯМР-моделях. Это говорит о том, что она все же вносит вклад в структуру, но не обязательно присутствует в любой момент времени. Далее можно рассмотреть длины водородных связей в РСА-структуре и их распределения в моделях ЯМР-структуры (таблица 1).
Таблица 1. Длины выбранных водородных связей в РСА-структуре и характеристики их распределения в ЯМР-моделях
| Связь | Положение | Расстояние между атомами (в ангстремах) | Встречаемость в моделях ЯМР (%) | |||
| РСА | ЯМР | |||||
| минимум | медиана | максимум | ||||
| A88-K91 | Петля на поверхности | 3.2 | - | - | - | 0 |
| К18-E35 | В глобуле белка между альфа-спиралью и бета-листом | 3.1 | 1.8 | 2.2 | 2.8 | 33 |
| E67-T71 | На поверхности белка в α-спирали | 2.8 | 1.8 | 2.1 | 2.5 | 100 |
Внимательно рассмотривая эту таблицу, можно сделать три наблюдения:
1. В структуре, полученной методом ЯМР, длины двух последних водородных связей больше, чем в
структуре, полученной методом РСА. Это можно объяснить двумя способами: во-первх, может быть, что,
из-за взаимодействия с соседними белками в кристалле, эти связи удлинились. И во-вторых (объяснение,
которое представляется мне более вероятным), это просто является признаком того, что в РСА-модели
эти расстояния могли быть не очень точно определены.
2. Водородная связь в α-спирале жестче, чем связь между α-спиралью и β-листом, то есть
дисперсия расстояний меньше.
3. Несмотря на то, что был выставлен порог длины водородной связи 3.5 Ангстрем, расстояние между атомами
в случае второй водородной связи (между α-спиралью и β-листом) принимает значения либо меньше 2.9 Ангстрем, либо больше 3.5. Это
может быть как случайным явлением, так и свидетельством того, что существует набор альтернативных
конформаций этих аминокислот, который делает распределение расстояний более дискретным. Специально
я этот факт не проверял.
Выводы
Несмотря на массу различий двух рассмотренных структур, можно сказать, что в общем структуры очень похожи,
и водородные связи, участвующие в образовании главных компонент структуры, наблюдаются и при разрешении
методом РСА, и при разрешении методом ЯМР. Причем водородная связь в элементе вторичной структуры -- α-спирали,
сохраняется во всех моделях ЯМР, в то время, как связь, поддерживающая третичную структуру, есть
не во всех моделях ЯМР. Водородная связь, наблюдаемая во внешней петле в РСА-структуре, не обнаруживается
в ЯМР-структурах. Это согласуется с выводами статьи
Крешимира Сикича с коллегами 2010 года, где говорится, что, во-первых конформации аминокислот в петлях
в растворе могут сильно отличаться от их конформаций в кристалле. А во-вторых, такой параметр, как
доступность растворителя, в кристаллах отличается от раствора. И, поскольку этот параметр
весьма важен для устойчивости водородной связи, логично, что в кристалле связь в петле образовалась, а
в растворе -- нет.
Также можно сравнить две структуры по формальным критериям, что позволяет сделать сервис
PDBeFold. Его выдача выглядит примерно так:
RESULT SUMMARY ## Q-score P-score Z-score RMSD Nalgn Nsse Ngaps Seq-% Nmd Nres-Q Nsse-Q Nres-T Nsse-T Query Target 1 0.7039 9.142 8.903 1.673 122 10 1 1 0 126 10 128 10 PDB 1chn:A PDB 1cey:A
Рассмотрим четыре параметра: Q-score, P-score, Z-score и RMSD:
Q-score дает информацию о качестве выравнивания Сα-атомов, принимая во внимание RMSD и длину
последовательности. Он равен 1, если структуры идентичны и очень бысто убывает с увеличением RMSD.
Значение 0.7 указывает на большое сходство последовательностей.
P-score это минус десятичный логарифм P-value, то есть вероятности того, что перед нами две случайные структуры.
Z-score это гауссова статистика, то есть такое значение, что интеграл стандартного нормального распределния
от него до бесконечности дает нам p-значение.
RMSD это дисперсия расстояния между Сα-атомами в структурном выравнивании. Значение 1.67 -- весьма хорошее,
так как в указанной ранее статье Сикича говорится, что RMSD между ЯМР и РСА-структурами колеблется между
1.5 и 2.5.
Из этого анализа можно сделать единственный вывод, что структуры очень похожи.