| Главная |
| Семестры |
| О себе |
| Ссылки |
7. Определение вторичной структуры
Для выполнения задания была взята структура с идентификатором 5DBQ, соответствующая
тиоредоксину. В ней имеется 4 α-спирали и 4 β-тяжа разной длины.
В отличие от третичной, вторичная структура предсказывается достаточно хорошо,
для ее предсказания написаны такие программы, как, например, DSSP и Stride.
Командами:
mkdssp -i 5dbq.pdb -o 5dbq.dssp
stride -h 5dbq.pdb -f5dbq.stride
они были запущены и было получено два файла с выдачей:
для DSSP и для Stride.
Точность предсказаний определялась по исходному PDB-файлу. Сравнение "настоящей" разметки
с разметками DSSP и Stride приведено в таблице 1.
Таблица 1. Сравнение разметок из PDB и полученных программами DSSP и Stride
| Элемент | PDB | DSSP | Stride | ||||
|---|---|---|---|---|---|---|---|
| Начало | Конец | Начало | Конец | Начало | Конец | ||
| 1 | Спираль | 7 | 18 | 8 | 17 | 8 | 18 |
| 2 | Спираль | 32 | 48 | 33 | 47 | 33 | 48 |
| 3 | Спираль | 63 | 70 | 64 | 69 | 64 | 70 |
| 4 | Спираль | 94 | 106 | 95 | 105 | 95 | 105 |
| 5 | β-тяж | 53 | 59 | 53 | 59 | 53 | 59 |
| 6 | β-тяж | 22 | 28 | 22 | 28 | 22 | 28 |
| 7 | β-тяж | 77 | 82 | 77 | 82 | 77 | 82 |
| 8 | β-тяж | 85 | 91 | 85 | 91 | 85 | 91 |
В принципе, предсказания получились хорошие, β-тяжи предсказываются немного лучше, чем α-спирали; программа Stride дает немного более близкие к исходной разметке предсказания, хотя для таких утверждений статистики явно недостаточно. Также следует отметить, что выдача Stride более удобна и проста для восприятия и парсинга.
8. Совмещение структур. 3D поиск
Поиск сруктурных гомологов белка
Для выполнения задания была выбрана структура белка крамбина с идентификатором 3NIR, для которой сервис PDBeFold находит 491 структурных гомологов. Из них было выбрано 4 не очень близких, но и не очень дальних гомолога:
1BHP:A Q=0.83 RMSD=0.83 %seq=32 2PLH:A Q=0.83 RMSD=0.87 %seq=32 3SZS:E Q=0.83 RMSD=0.99 %seq=31 2EYC:A Q=0.67 RMSD=1.81 %seq=93
Выравнивание структурных гомологов
Эти структуры были использованы для построения структурного выравнивания и выравнивания последовательностей (рис. 1, рис.2).
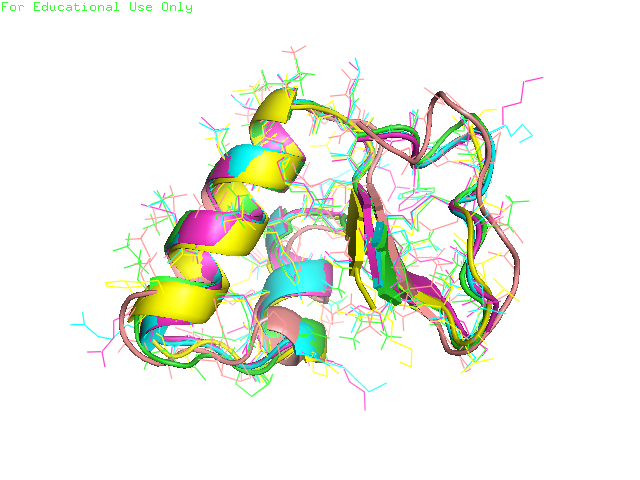
Рисунок 1. Структурное выравнивание выбранных последовательностей.

Рисунок 2. (Сверху) выравнивание последовательностей выбранных структур алгоритмом MUSCLE. (Снизу) Выравнивание, полученное по структуре.
Сравним два полученных выравнивания (рис. 2). Можно заметить, что они очень похожи, но у структурное выравнивание менее блочное. Обратим внимание на колонку с номером 20. В структурном выравнивании там стоят аргинины, глицины и пролин, которые действительно расположены близко в пространстве в пространстве (рис. 3). В выравнивании последовательностей, эта колонка занята исключительно глицинами. Теперь посмотрим на расположение этих остатков в пространстве (рис. 3).

Рисунок 3. Расположение двадцатых глицинов (выделены серым) на структурном выравнивании.
Из этого рисунка видно, что глицины образуют начало петель, соединяющих альфа-спирали, что и является частой фунцией глицина в структуре. Этот факт, а также то, что структурное выравнивание на этом участке не позволяет сделать однозначный вывод, наводит на вывод, что в данном случае лучше сработало выравнивание последовательностей.
Сравнение гибкого и жесткого выравнивания
В статье, описывающей алгоритм FATCAT, говорится, что в среднем гибкое совмещение дает выравнивание большей длины. Одним из ярчаших примеров этого преимущества является пара структур 1AJ3 и 2SPC, отличие которых заключается в одной мутации в середине альфа-спирали 2SPC, которая создает в этом месте петлю. В результате, жесткое совмещение не выравнивает половину этой спирали, в то время как гибкое с этой задачей справляется. Сервис PDBeFold дает Z-score выравнивания 2.5 и P-value 0.006, в то время, как FATCAT дает P-value 1.93e-06, что гораздо лучше. Причиной, может являться то, что FATCAT вносит петлю в середину длинной альфа-спирали и сгибает ее, улучшая таким образом качество выравнивания.
9. Гидрофобные кластеры
Поиск гидрофобных кластеров
С помощью сервиса CluD можно находить гидрофобные кластеры в структуре белка. Рассмотрим структуру белка крамбина с идентификатором 3NIR. Известно (см. практикум 1), что крамбин -- это плотный белок, единственной функцией которого является образование плотных кристаллов, в которых запасается энергия и аминокислоты. Поэтому можно ожидать, что в крамбине будет всего один гидрофобный кластер. Меняя параметр порогового расстояния в сервисе CluD, Можно изучить поподробнее свойства ядра крамбина (рис. 4).

Рисунок 4. Гидрофобные кластеры в крамбине при разных значениях параметра порога расстояния.
Как мне кажется, наиболее правдоподобные картины получились при значениях 5.4 и 6.5 Ангстрем. При больших значениях частью кластера становятся сильно отдаленные аминокислоты участков, явно выступающих в раствор, а при меньших значениях кластеры распадаются на мелкие части, которые визуально должны быть связаны. Визуально кажется, что значение в 6.5 Ангстрем оказалось удачнее параметра по умолчанию, так как бета-лист, очевидно входящий в центральный гидрофобный кластер, при таком пороге не разделяет его на две части.
Гидрофобные кластеры в структуре взаимодействия ДНК-белок
Теперь посмотрим на гидрофобные кластеры в комплексе ДНК с белком. Для выполнения задания была использована уже рассматривавшееся структура 3HDD.

Рисунок 5. Гидрофобные кластеры в структуре 3HDD при разных значениях параметра порога расстояния.
Можно заметить, что в данном случае не наблюдается какой-то специальной гидрофобной области контакта белка с ДНК. Впрочим, контакт достаточно плотный, и при повышении порога кластеры ДНК и белка сливаются.
10. Поверхность белка
Для выполнения задания была выбрана структура с идентификатором 1QP0, отвечающая операторному комплексу палиндромного гипоксантинового рецептора кишечной палочки. В начале, с помощью операций симметрии, была восстановлена "вторая половина" PDB-файла, то есть была воспроизведена биологическая единица. На рисунке 6 Показана поверхность контакта мономеров белка в активном димере.

Рисунок 6. Поверхность контакта мономеров белка в активном димере пуринового репрессора.
На рисунке 7 показана поверхность контакта белка с ДНК и на рисунке 8 показана та же картина, только молекула ДНК изображена в форме палочек (sticks).

Рисунок 7. Поверхность контакта пуринового репрессора c ДНК.

Рисунок 8. Поверхность контакта пуринового репрессора c ДНК.

Рисунок 9. Поверхность контакта мономеров белка в активном димере пуринового репрессора. Красным отмечены области, входящие в гидрофобные кластеры.
Далее была запущена программа CluD с параметрами максимального расстояния 5.4 Ангстрема и минимального размера кластера 10 атомов. Таким путем было найдено 6 кластеров в одном мономере. С помощью скрипта, переводящего скрипты RasMol в скрипты PyMOL, была сгенерирована последовательность команд, выделяющих гидрофобные кластеры, найденные CluD, в белке. На рисунке 9 изображена поверхность контакта мономеров, раскрашенная по принадлежности к гидрофобным кластерам. Можно заметить гидрофобные контакты между мономерами вблизи ДНК.
11. Структурная классификация
Для выполнения задания была выбрана структура с идентификатором 1L7V, и для нее был проведен поиск границ доменов в четырех базах: SCOP, PFAM, ECOD и CATH. Результаты Для одного из доменов -- укладки АВС-транспортера типа II, представлены в таблице 2.
Таблица 2. Границы домена АВС-транспортера типа II в базах SCOP, PFAM, ECOD и CATH
| База данных | Найденные домены | Границы доменов |
| Pfam | PF01032 | 22-323 |
| SCOPe | d1l7va | 1-324 |
| CATH | 1l7vB00 | 2-324 |
| ECOD | e1l7vA1 | 1-324 |
Можно заметить, что базы SCOP, ECOD и CATH дают практически идентичный результат, в то время, как начало домена согласно базе Pfam сдвинуто в сторону середины белка. Однако в целом все базы дают очень похожие результаты.
12. Поиск в PDB
Возможности Advanced Search
Был сформулирован запрос в Advanced search базы данных PDB на структуры, разрешенные РСА с разрешением от 2.5 до 5 Ангстрем. Также структуры должны были иметь свободные лиганды, быть дикого типа (не мутанты), иметь длину между 100 и 150 аминокислот и принадлежать кишечной палочке. Полный текст запроса:
Resolution is between 2.5 and 5.0 and Ligand Search : Has free ligands=yes and Percent Sequence Alignment Search : PDB can contain Expression Tag sequence = Yes , and Sequence Length is between 100 and 150 and Taxonomy is just Escherichia coli (E. coli)
Далее, с помощью опции custom report, была сгенерирована Excel-таблица с результатами поиска. Также был скачен файл со всеми последовательностями и файл с идентификаторами.
Таблица филаментов
Далее, опять же с помощью опции Advanced search, был получен список структур филаментов, определенных методом электронной микроскопии. Текст запроса:
Text Search for: filament and Holdings : Molecule Type=ignore Experimental Method=ELECTRON MICROSCOPY
Далее, фильтрацией записей в Custom report, был получен файл, содержащий записи только о белках со сходством выше 50% остатков.