Сигнал инициации репликации в ориджине OriL в митохондриальной ДНК.
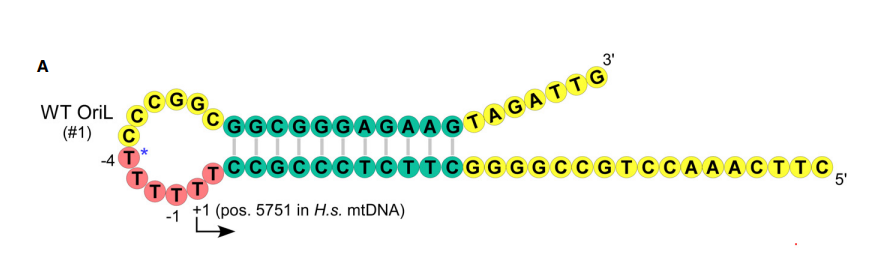
В митохондриальном геноме роль праймазы выполняет белок, который осуществляет и транскрипцию - POLMRT (mtRNAP). Сигнал представляет собой шпильку на тяжелой цепи (H-цепь) митохондриальной
ДНК в области ориджина OriL (рисунок 1). Данная шпилька образуется во время асинхронной репликации мтДНК. Когда реплисома на легкой цепи (L-цепь) мтДНК проходит участок L-цеппи напротив которого находится OriL
она осталяет данный участок H-цепи в одноцепочечном состоянии, что приводит к образованию stem-loop (шпильки). Данная шпилька и является сигналом, который адресован POLMRT и ДНК-полимеразе гамма. Первым к стержню шпильки присоединяется Polg, затем
к петле присоединятся POLMRT. Большая часть петли состоит из скользкой последовательности, состоящая из тимидиловые нуклеотидов.
Второй нуклеотид от 3'-конца данной последовательности является сайтом начала транскрипции.
Данная последовательность позволяет mtRNAP создавать гомополимерный
транскрипт путем последовательного включения остатков AMP без необходимости транслокации POLMRT вдоль ДНК-матрицы. Процесс проскальзывания продолжается до тех пор, пока не будет сгенерирован по меньшей мере гибрид РНК–ДНК в количестве 5-6 п.н. Учеными было доказано,
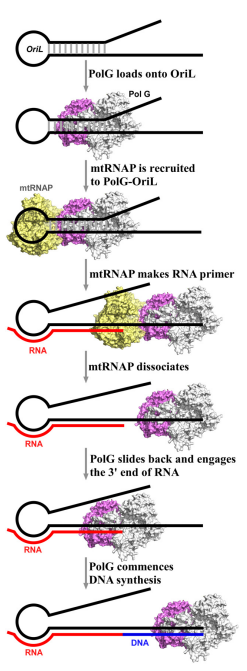
что данный гибрид играет ключевую роль в стабильности комплекса mtRNAP-oriL, что позволяет POLMRT вперед против PolG, заставляя последний скользить вниз по ДНК-матрице.
Транскрипция с помощью mtRNAP продолжается до тех пор, пока размер зарождающегося гибрида РНК–ДНК не сыграет важную дестабилизирующую роль и не приведет к диссоциации mtRNAP. Когда mtRNAP диссоциирует, PolG скользит назад и захватывает 3’-конец праймера РНК, инициируя репликацию ДНК (рисунок 3).
Рисунок 1. Схема строения шпильки.Рисунок 2. Модель инициации репликации в OriL.
Позиционная весовая матрица для последовательности Козак человека.
Чтобы построить позиционную весовую матрицу для последовательности Козак человека, были выбраны 100 случайных генов, для которых известны координаты стартовых ATG. Для этого мною был написан скрипт positive.py, который принимает
на вход таблицу с информацией о генах человека human-genes.tsv и создает два файла:
learn_genes.txt содержит выравнивание без гэпов 100 нуклеотидных последовательностей, каждая из которых - 7 нуклеотидов до ATG + ATG + 3 нуклеотида после ATG (обущающая выборка последовательностей Козак). С помощью этой выборки будет строится PWM.
test_genes.txt содержит выравнивание без гэпов 300 нуклеотидных последовательностей, каждая из которых - 7 нуклеотидов до ATG + ATG + 3 нуклеотида после ATG (тестовая выборка последовательностей Козак)
Далее был написан скрипт negative.py который отобрал 300 последовательностей внутри случайных генов (то есть по сути нет оснований считать что ATG-окресность кодирует начало трансляции) и выдает следующий файл:
negative_test_genes.txt содержит выравнивание без гэпов 300 нуклеотидных последовательностей, каждая из которых - 7 нуклеотидов до ATG + ATG + 3 нуклеотида после ATG (выборка негативного контроля последовательностей Козак)
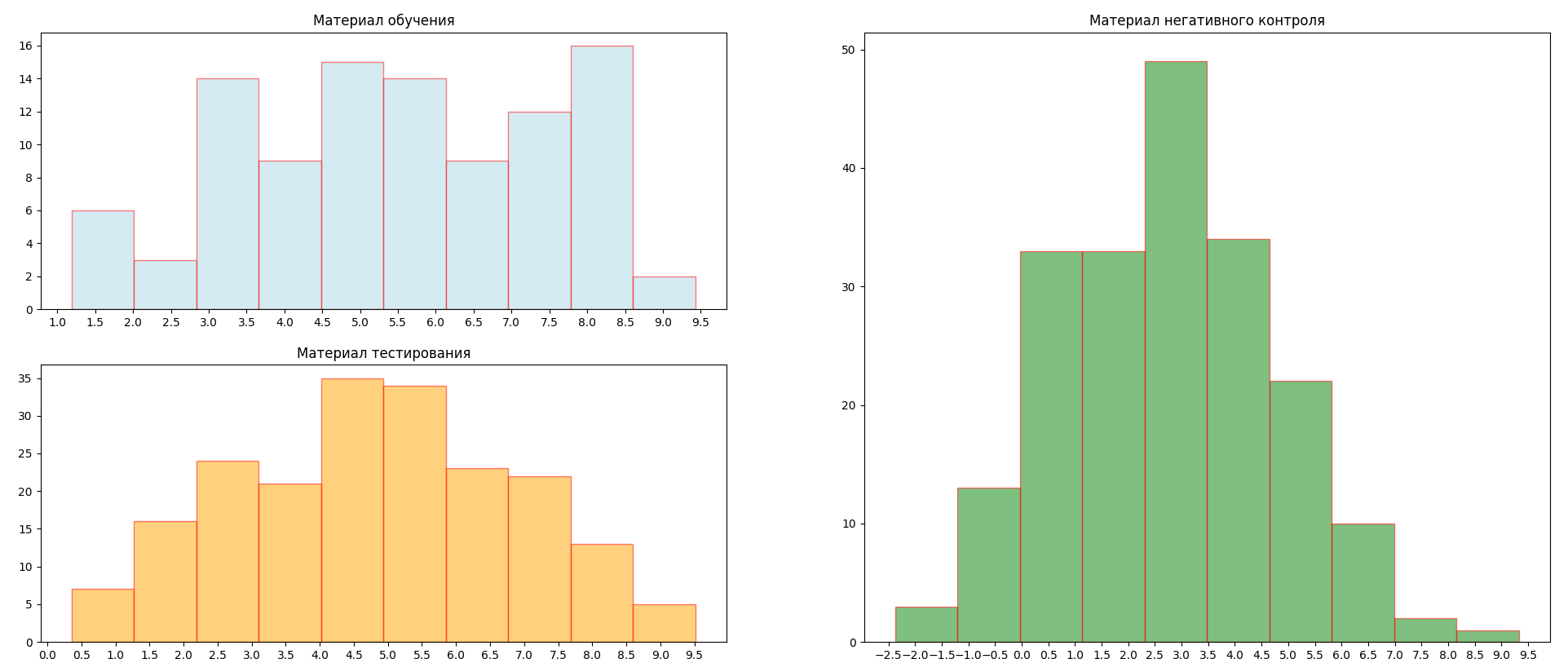
После этого была создана позиционная весовая матрица (таблица 1) и с помощью данной матрицы вычислены веса последовательностей каждой выборки (GC=41%, pseudocounts=0.1). Гистограммы распределения весов последовательностей представлены на рисунке 3 (Для осуществления данных действий также был мною написан скрипт pwm_build.py). По данным гистограммам можно
примерно оценить порог веса при котором мы считаем что последовательность содержит последовательность Козак. Пусть пороговое значение будет равно 4-м.
Таблицa 1. PWM.
1
2
3
4
5
6
7
8
9
10
11
12
13
A
-0.249
-0.612
-0.612
-0.017
0.4
0.081
-0.899
1.218
-5.691
-5.691
-0.389
0.112
-0.166
C
0.238
0.198
0.535
0.59
-0.623
0.414
0.668
-5.327
-5.327
-5.327
-0.13
0.668
-0.025
G
0.563
0.764
0.158
0.158
0.506
-0.187
0.476
-5.327
-5.327
1.582
0.931
-0.455
0.668
T
-0.676
-0.676
-0.166
-1.082
-0.986
-0.389
-0.676
-5.691
1.218
-5.691
-1.082
-0.745
-0.676
Рисунок 3. Гистограммы распределения весов. Голубой цвет - обущающая выборка, оранжевый цвет - тестовая выборка, зеленый цвет - выборка негативного контроля.
Обучающая выборка
Тестовая выборка
Выборка негативного контроля
Cигнал(+)
77
132
52
Cигнал(-)
23
68
148
Таблица 2. Таблица с количеством последовательностей, отобранных по порогу в трех выборках.
Далее было определено количество положительных и отрицательных сигналов в каждой из трех выборок по порогу (результат в таблице 2). Несмотря на то, что
в обучающей и тестовой выборках нашлись последовательности с отрицательным сигналом, процент положительных сигналов в обущающей выборке и тестовой выборке равен 77% и 68% соответственно, в то время как
для выборки негативного контроля данный процент равен 26, что говорит нам о том что данный порог неплохо определяет положительный сигнал.
Информационное содержание последовательности Козак H.sapiens
В данном разделе с помощью обучающей выборки и скрипта ic_builder.py была построена матрица информационного содержания последовательности Козак (таблица 3). Также с помощью сервиса
WebLogo3 был построен Logo (рисунок 4). Из схемы видно, что позиции 2,4,5,7-12, имеют значимый информационный вес, т.е.
есть основания считать, что данные выравненные последовательности обладают специфической функцией.
Таблицa 3. Матрица информационного содержания IC для последовательности Козак H.sapiens.
1
2
3
4
5
6
7
8
9
10
11
12
13
A
-0.083
-0.141
-0.141
-0.007
0.254
0.038
-0.156
1.761
0.0
0.0
-0.112
0.053
-0.06
C
0.089
0.072
0.27
0.315
-0.099
0.185
0.386
0.0
0.0
0.0
-0.034
0.386
-0.007
G
0.292
0.485
0.055
0.055
0.248
-0.046
0.227
0.0
0.0
2.286
0.698
-0.085
0.386
T
-0.146
-0.146
-0.06
-0.156
-0.157
-0.112
-0.146
0.0
1.761
0.0
-0.156
-0.151
-0.146
IC(j)
0.152
0.270
0.124
0.207
0.246
0.065
0.311
1.761
1.761
2.286
0.396
0.203
0.173
Рисунок 4. Logo, построенное на основе выравнивания последовательностей с сигналом.
Подсчёт числа сайтов GAATTC в полном геноме E.coli.
Для выполнения этого задания был использован геном штама E.coli rl0044.
С помощью скрипта было определено, что в геноме e.coli 712 сайтов GAATTC, а длина всего генома - 5256017 нуклеотидов. Также были определены частоты для
каждого нуклеотида. Для получения вероятности появления данного сайта в геноме, частоты нуклеотидов входящих в GAATTC были перемножены между собой - 0.000237. Помножив вероятность на длину генома, получим
математическое ожидание количества сайтов равное 1244.77, что почти в два раза больше реального количества сайтов в последовательности. Для оценки достоверности данного отличия, был проведен обычный односторонний Z-test. Можно считать что количество данных сайтов в геноме
имеет биномиальное распределение. В виду большой длины генома можно считать что случайная величина (количество сайтов в геноме) имеет нормальное распределение (по центральной предельной теореме) с математическим ожиданием равным 1224.77 и среднеквадратичным отклонением равным 35.3.
Примем за нулевую гипотезу то что среднее количество данных сайтов в геноме равно математическому ожиданию, а за альтернативную гипотезу, то что среднее меньше математического ожидания. Z-test показал p-value равное 8.48*10^-52. При таком маленьком значении мы можем отвергнуть нулевую гипотезу
и принять альтернативную при любом адекватном уровне значимости. Это говорит нам о том, что отличие достоверно