CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
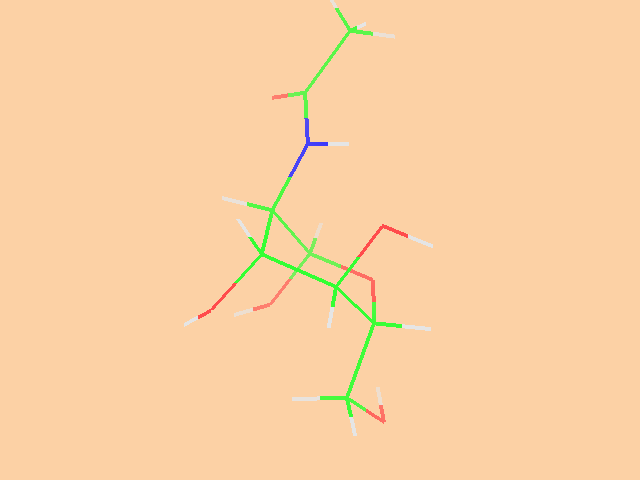
А так выглядит трехмерная структура NAG
Для начала, с сайта банка pdb нужно получить smiles NAG:
CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
А так выглядит трехмерная структура NAG
Сначала нужно определить координаты центра масс в PyMol, создав для этого псевдоатом. Координаты вставляем в файл vina.cfg .
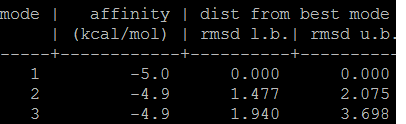
Получены следующие значения энергии и геометрической разности наилучших состояний:

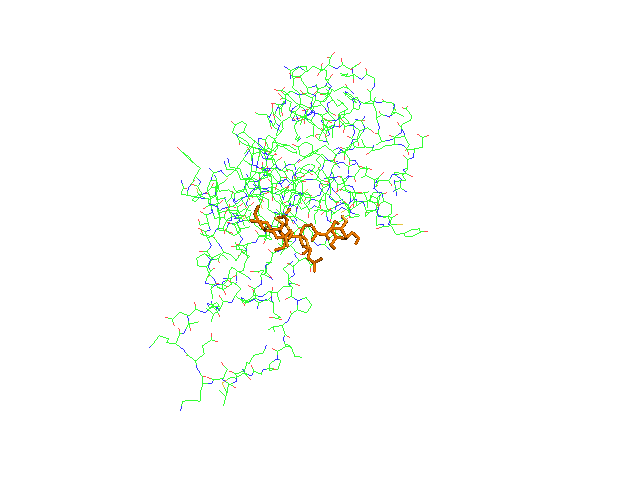
Если совместно открыть файл с белком и полученный nag_prot.pdbqt (лиганд выделен оранжевым):
Получены следующие значения энергии и геометрической разности наилучших состояний:
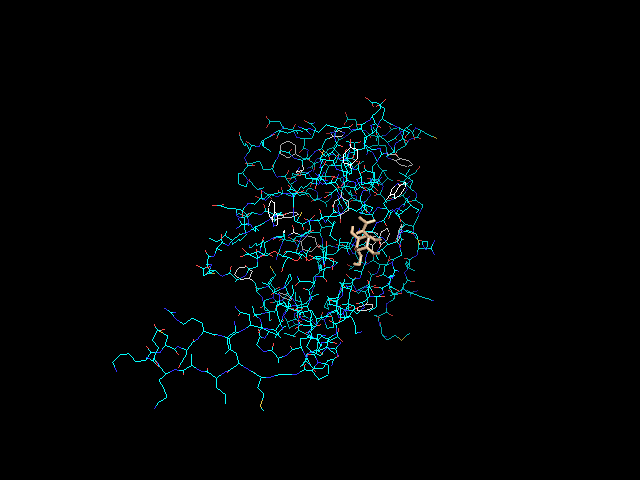
Если совместно открыть файл с белком mod4_rigid.pdbqt и полученный nag_out.pdbqt (лиганд выделен оранжевым):
Met -> H: C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
Met -> NH2: C(N)(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
Met -> OH: OC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
Met -> PH:C1=CC=CC=C1C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
Met -> H
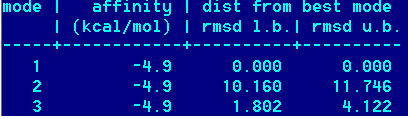
Met -> OH
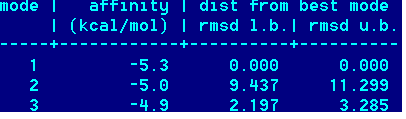
Met -> NH2
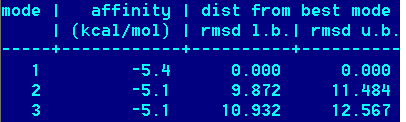
Met -> PH
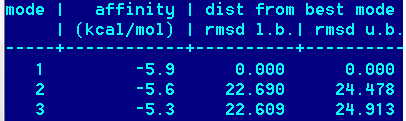
Как видим, наилучшие значения афинности получены при замене на Н (лучше чем при СН3).
Met -> OH
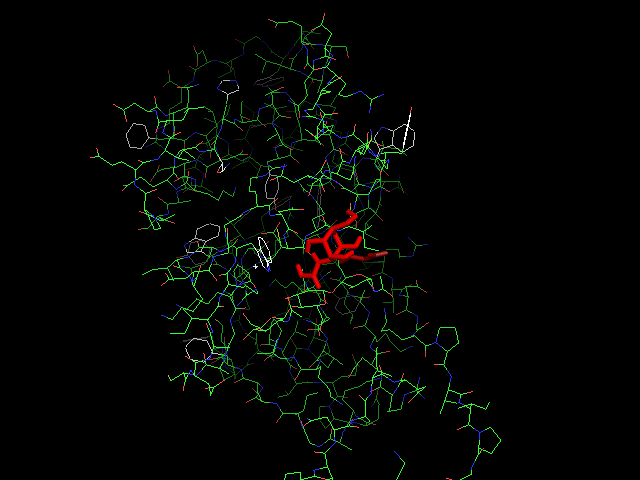
Met -> NH2
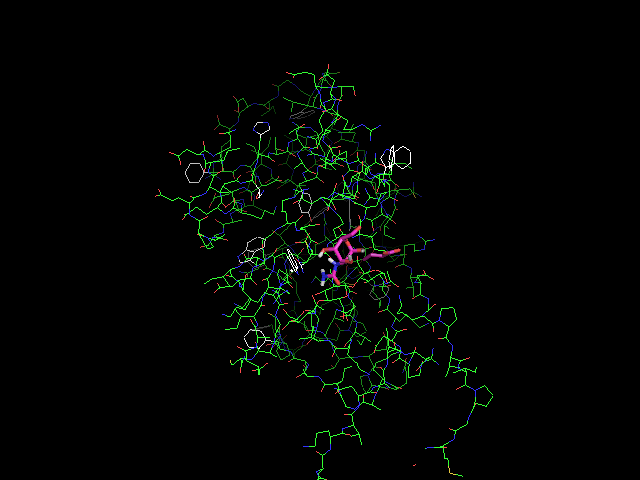
Met -> PH
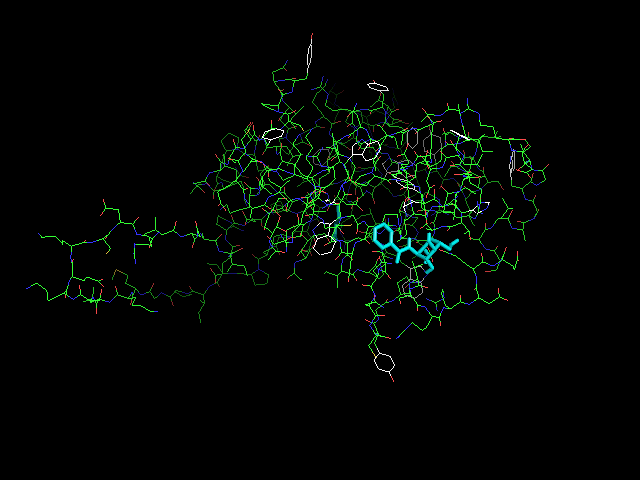
Наиболее компактно встроился лиганд с аминогруппой.