Сравнение выравниваний
1. Сравнение выравниваний одних и тех же последовательностей разными программами
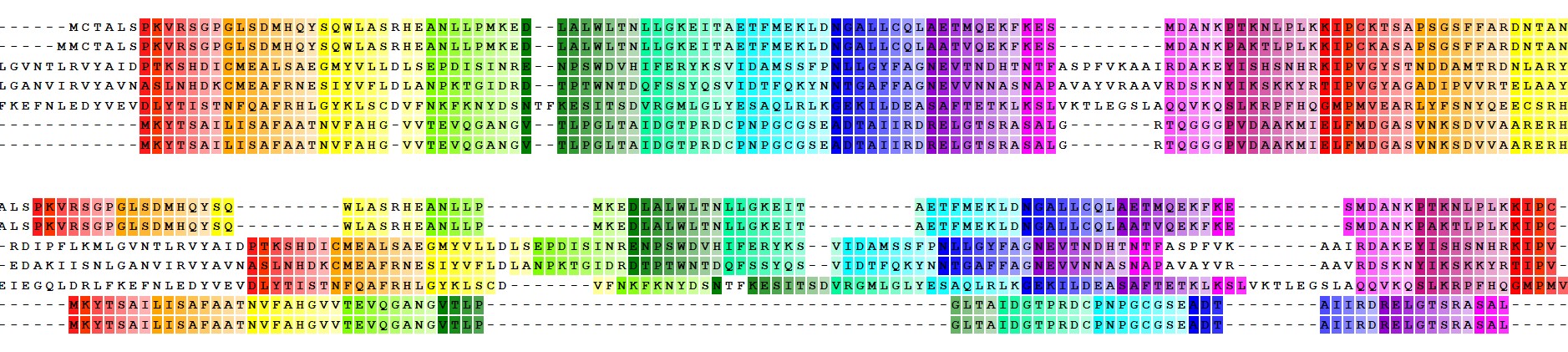
Для выравнивания я взяла мнемонику KITH (из практикума 9) и GAS2. Для сравнения вывода я взяла программы MUSCLE и Clustal.
kith_human, kith_mouse, kith_hhv1, kith_dictd, kith_staan, kith_ecoli, kith_bacsu
Выравнивания программ очень похожи. Вероятно, это связано с тем, что мало гэпов, из-за которых обычно происходит смещение. Например,
участок с 91 по 97 (для обеих последовательностей) отличается большим количеством гэпов, поэтому программы по-разному расположили глицин
относительно большого инделя. Или участок с 224 по 232: программа MUSCLE нашла целый блок на этом участке, где много похожих аминокислот у
наших белков, в то врем как Clustal не показывает никакой гомологичности конечных участков более коротких белков в наших выравниваниях.
| MUSCLE | Clustal | Длина участка |
|---|---|---|
| 18-64 | 18-64 | 47 |
| 67-90 | 67-90 | 24 |
| 103-175 | 103-175 | 73 |
Я думаю, что мы добились большого сходства между белками только за счет того, что было мало гэпов, потому что в начале и конце выравниваний,
где сильно разнятся последовательности, результаты работы программ были разными.
GAS2_HUMAN, GAS2_MOUSE, GAS2_YEAST, GAS2_SCHPO, GAS2_HELAN, GAS2_MAGO7, GAS2_MAGOR
Для этих белков программы сработали совершенно по-разному, если в качестве референсной брать выдачу Clustal, то сервис VerAlign показывет, что
нет ни одной полностью сходящейся колонки, даже если верхние 4 последовательности совпадают, то нижние "уезжают" из-за гэпов. Если же в качестве
референсной использовать выдачу MUSCLE, то ситуация становится намного хуже. Таким образом, я не могу найти одинковых кусков в данных выравниваниях.
Сравнение выравниваний с помощью программы Елизаветы Плешко:
Результат сравнения выравниваний для мнемоники kith записан в файл.
Доля одинаково выравненных позиций в первом выравнивании: 75%
Доля одинаково выравненных позиций во втором выравнивании: 75%
Результат сравнения выравниваний для мнемоники GAS2 записан в файл.
Доля одинаково выравненных позиций в первом выравнивании: 1%
Доля одинаково выравненных позиций во втором выравнивании: 1%
2. Построение выравнивания по совмещению структур
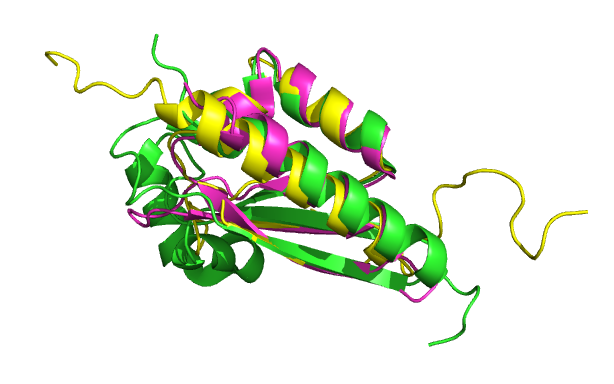
Для выравнивания белков в PyMol я взяла 5els, 2ctm, 4lij из семейства KH_1 (PF00013). К сожалению, PyMol
при парном выравнивании не выдает последовательности, поэтому я не смогла их ни с чем сравнить. Однако точно могу сказать,
что пространственные структуры довольно хорошо сопоставились.
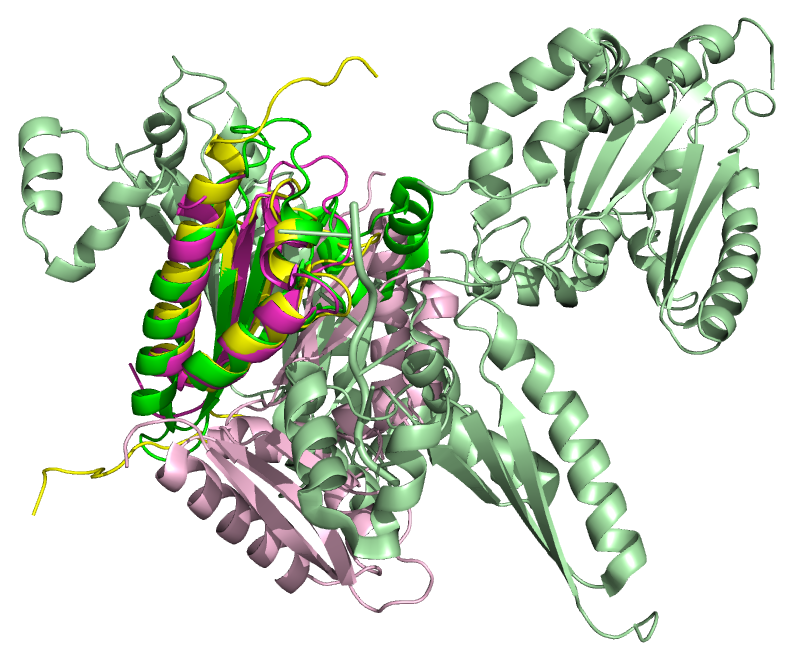
Я решила также сделать выравнивание трех последовательностей с помощью Clustal и webPRANK, но результаты
оказались очень странными... Здесь можно скачать выравнивание (Clustal слева и webPRANK справа).
Вероятно, это свезано с тем, что один белок состоит из одной цепи, а остальные из трех и шести. Хотя все равно результат странный.
3. Описание программы T-Coffee
T-Coffee - это программа множественного выравнивания, которая позволяет комбинировать уже полученные в других программах
выравнивания с получением нового, максимально хорошо согласующегося со всеми методами. Для начала он попарно сравнивает последовательности,
и только после этого получает полное выравнивание, а также серии локальных выравниваний. Впоследствии из всех этих выравниваний получается
множественное выравнивание.
Однако эта программа может как и все остальные выравнивать обычные последовательности:
1. Изучает библиотеку на наличие участков, которые могут быть выровнены
2. Считает вес каждого из них
3. Комбинирует их так, чтобы получилось множественное выравнивание, в котором окажутся участки, имеющие больший суммарный вес
Эта программа также позволяет самостоятельно выровнять определенные важные участки и задать им больший вес.
Для вашего удобства T-Coffee может сгенерировать (по умолчанию) свой собственный список, выполнив все возможные глобальные попарные
выравнивания и 10 наилучших локальных выравниваний, связанных с каждой парой последовательностей. Каждая пара остатков, наблюдаемых
выровненными в этих попарных выравниваниях, становится строкой в библиотеке.