Корова! Продолжение.
1. Описание фрагмента ДНК
Для поиска CDS я решила искать гены в NCBI graphics, используя записи в protein.faa. Случайным
образом выбрала белок "cilia- and flagella-associated protein 20"* (ID: NP_001003905.1).
В поисковую строку Genome Data Viewer занесла ">NP_001003905.1 cilia- and flagella-associated protein 20".
Расположение гена CFAP20: (25.995.000; 26.008.254)
Длина гена: 13,255 нп
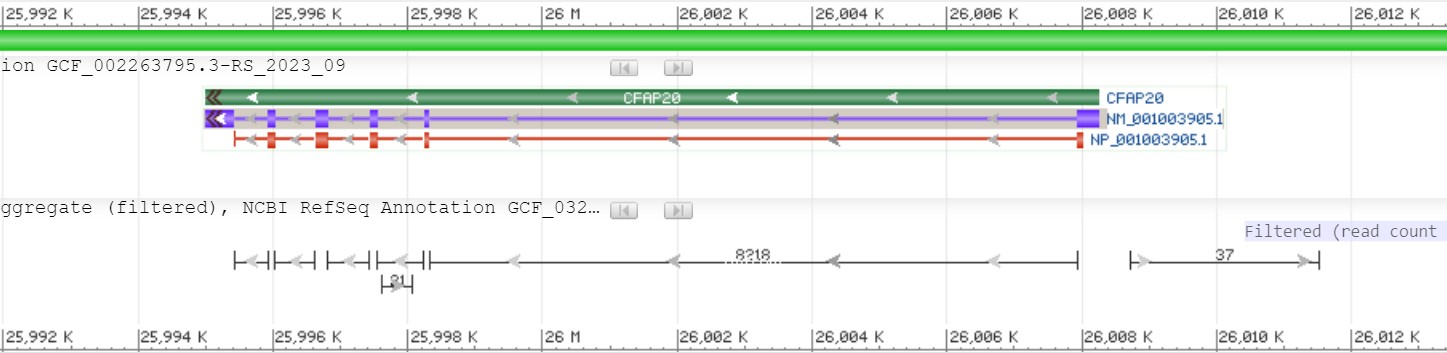
Зелёным цветом обозначен ген CFAP20 (ID: NC_037345.1), его длина 13,255 нп.
Фиолетовым - мРНК NM_001003905.1, её длина: 1,332 нп.
Красным - CDS NP_001003905.1 (только одна в этом гене), длиной 582 нп, а белок, кодирующийся ею имеет длину 139 ак.
Стрелочками внизу показаны интроны.
2. Исследование алгоритмов BLAST
Мое животное находится в классе Mammalia, поэтому я решила взять в соседним с нам классе Sauropsida класс Lepidosauria
(Taxonomy ID: 8504): они вместе входят в надкласс Tetrapoda и кладу Amniota. В класс Lepidosauria входят отряды: клювоголовые и чешуйчатые.
На самом деле сначала я решила выбрать в качестве таксона сравнения Sauropsida, но при применении поиска алгоритмами BLAST получалось
больше 250 результатов, так как таксон включает в себя не только рептилий, но и птиц. По этой же причине я решила для всех алгоритмов поставить
1000 выдач.
megablast:
я решила провести сравнение выдач при длине слова в 32 нуклеотид и 28. В первом случае мы получали 140 последовательностей с процентом покрытия от 0 до 8 (мода = 3%), процент сходства от 75 до 95 (мода: примерно 81%), что как по мне очень достойно (!) значения e-value стремятся к нулю. Во втором случае 225 результатов, процент покрытия так же от 0 до 8, но в этот раз очень много нулей, как и любого другого значения (все примерно поровну), процент сходства от 77 до 94, мода немного выше, чем в первом случае, значения e-value стремятся к нулю. Наверное, лучше все же использовать длину слова 28, чтобы было больше находок, среди которых будут последовательности с большим сходством, отсеченные при более большой длине слова. Стоит использовать, если хочется найти гены либо у близких групп, либо высоко консервативные, так как мы на примере убедились, что небольшое увеличение длины легко отсекает много вариантов, которые могли быть даже лучше найденных, но расположение мутаций не позволило их отобрать сразу.blastn:
при использовании слова из 11 нуклеотидов выдача состояла из 3586 результатов. Сначала меня это испугало, но затем я полистала и поняла, что результаты очень странные. Часть - встречи в разных хромасомах одного и того же вида, а 99% это предсказанные мРНК. процент покрытия редко превышал 4, а сходство было на уровне 70-80%. Данную программу можно применять чтобы узнать в каких организмах тоже есть такой ген.blastx:
использовалось слово длиной 5 нуклеотидов. Выдача состояла из 728 содержательных записей, ничего predicted почти не было. Покрытие все еще достаточно маленькое, ниже 5 в основном, процент сходства в районе 75, но бывают и выбивающиеся значения около 36%, что странно. С помощью этого алгоритма получится хорошо отследить эволюцию белков, так как происходит трансляция гена и поиск по белковой базе данных, а значит синонимичные замены не будут отвлекать от сути.tblastx:
этот прелестный алгоритм впринципе не захотел работать ни при каких условиях, даже при уменьшении выборки до 10 результатов.... Незаменим в ситуациях, когда нужно отследить эволюцию белков, но организм редкий и его белок не занесен в белковую базу данных, ведь у этого алгоритма поиск происходит по автоматически созданным последовательностям полипротеинов.3. Использование blastn на kodomo
Для индексирования последовательности была использована команда:
makeblastdb -in ~/term3/NC_037345.1.fa -dbtype nucl
Для запуска алгоритма blastn использовались команды:
blastn -task blastn -evalue 10 -query ecoli23.txt -db NC_037345.1.fa -out blastn_23.fna -outfmt 7 -word_size 4
blastn -task blastn -evalue 10 -query ecoli16.txt -db NC_037345.1.fa -out blastn_16.fna -outfmt 7 -word_size 4
Сначала выдача была нулевой, поэтому пришлось поднять e-value до 10, тогда все получилось, также я решила длину слова уменьшить до 4,
так как времени мне не жалко, а хочется все же увидеть результат, состоящий не из одной строки. Параметр -outfmt и значение 7 означали,
что в качестве выходного фацла я хочу увидеть таблицу. Скачать файлы с выдачей вы можете ниже:
Мы проводили поиск по некодирующим последовательностям рРНК кишечной палочки, 16s (в малой субъединице рибосомы) и 23s (в большой) рРНК.
Я думаю, что мы взяли именно их, потому что они имеют важное значение в клетке, соответственно, можно будет найти их гомологов.
Так как РНК у нас была некодирующая, то выбор стоит между алгоритмами, которые ищут результат в нуклеотидных базах данных. То есть это
megablast или blastn. Но очевидно, что бактериии и эукариоты очень далеки, поэтому надо использовать программу, которая способна работать с
сильно различающимися последовательностями, поэтому blastn.
Для каждой рРНК выдача содержала 10 последовательностй. Так как я не меняла значение, то по умолчанию их могло мне показаться 500, так
что мы можем увидеть все последовательности.
Я так понимаю, что для каждой рРНК у меня вся выдача состояла из копий гена этой рРНК у одного и того же организма. Результат получился не очень..(
Аннотацию найти не удалось.
*Последовательность аминокислот белка, кодирующегося геном "NP_001003905.1":
MFKNTFQSGFLSILYSIGSKPLQIWDKKVRNGHIKRITDNDIQSLVLEIEGTNVSTTYITCPADPKKTLG
IKLPFLVMIIKNLKKYFTFEVQVLDDKNVRRRFRASNYQSTTRVKPFICTMPMRLDDGWNQIQFNLSDF
TRRAYGTNYIETLRVQIHANCRIRRVYFSDRLYSEDELPAEFKLYLPVQNKAKQ