Учебный сайт Кузнецовой Марии
Эволюционные домены
Выбор домена
Для данной работы выбрала MipZ - АТФаза, образующая комплекс с белком ParB, разделяющим хромосомы вблизи ориджина репликации. Он отвечает за временную и пространственную регуляцию образования FtsZ кольца. Pfam AC PF09140, Pfam ID MipZ. Домен входит в 431 последовательность из 400 организмов, входит в 7 доменных архитектур, в базе данных PDB находится 5 трехмерных структур. Ниже приведено изображение каталитической субъединицы, содержащий данный домен (рис. 1). Страница домена в Pfam.
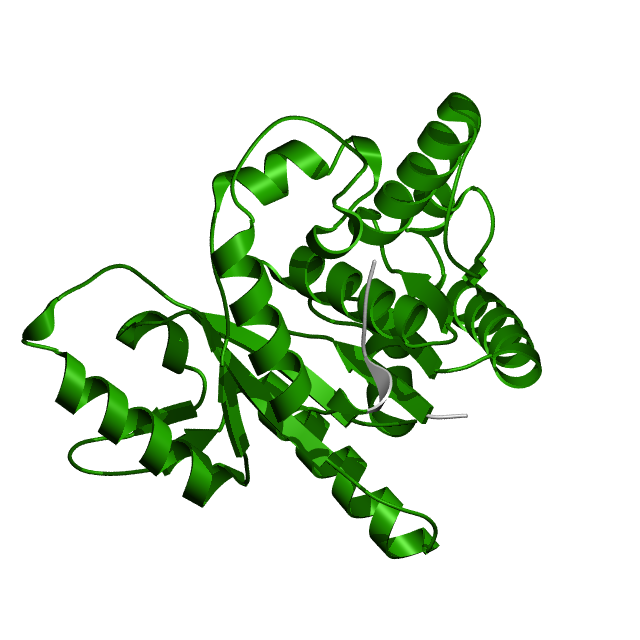
Рисунок 1.АТФаза MipZ, пространственная структура.
С помощью Jalview загрузила выравнивание домена. Ассоциировала структуру 2XJ9 c Q9A6C9_CAUCR. Сделала раскраску по ClustalX и консервативность 13%. Сохранила выравнивание в формате .fasta и .jar.
Выбрала три доменные архитектуры, которые показались мне наиболее интересными, для дальнейшего изучения - белок с одним доменом MipZ (PF09140); DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609); MipZ (PF09140) + ParA (PF10609). Это наиболее "популярные" доменные архитектуры, их 175, 127 и 118 последовательностей соответственно. PF10609 - ParA/MinD ATФаза-подобный домен, DUF59 (PF01883) - домен с неизвестной функцией, находится в цитоплазме многих организмов. Остальных доменных архитектур всего 4, а последовательностей из них 11 штук, поэтому описывать их не будем. Ссылка на список архитектур в Pfam.



Получила таблицу с информацией об архитектуре всех последовательностей, содержащих выбранный домен, с помощью скрипта swisspfam_to_xls.py. Составила список последовательностей с указанием доменной архитектуры. Использовала сводную таблицу в Excel: строки – AC последовательностей, столбцы – домены Pfam. Скачала полные записи всех последовательностей из Uniprot, запустила скрипт uniprot_to_taxonomy.py. Перенесла таксономию с основную таблицу. Далее в качестве таксона выбрала все клеточные организмы, а в качестве его подтаксонов - Archaea и Bacteria.
Выбрала по 25 последовательностей из каждой выбранной выше архитектуры. С помощью скрипта filter_alignment.py оставила только выбранные последовательности из выравнивания. Далее открыла проект в JalView, удалила пустые колонки, создала группы по архитектурам (сверху находятся последовательности, взятые из белков с одним доменом, внизу - последовательности в архитектуре), удалила плохо выровненные участки. Сохранила выравнивание в формате .jar. Ниже привожу изображение выравнивания (ClustalX, консервативноcть 30) (рис. 2). Участки, соответствующие альфа-спиралям и бета-тяжам, определены примерно, потому что выравнивание после удаления плохо выровненных участков значительно уменьшилось в длине, и остались только намеки на вторичные структуры.
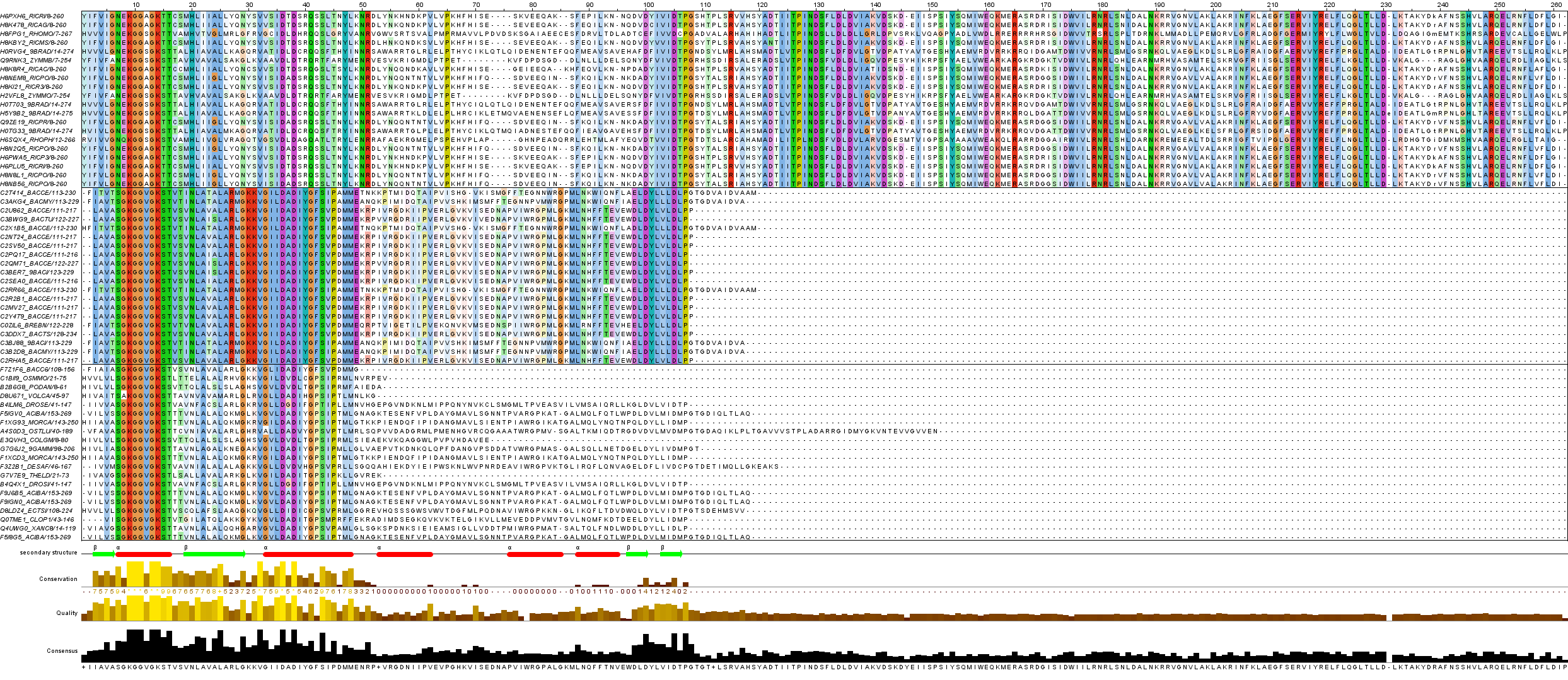
Рисунок 2.Выравнивание отобранных последовательностей домена, сверху группа c одним доменом MipZ (PF09140); далее DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609); MipZ (PF09140) + ParA (PF10609).
Исходя из выравнивания, все-таки можно понять, что большая консервативность в значимых участках - альфа-спиралях и бета-тяжах - существует. Наибольшая консервативность наблюдается у трехдоменных белков. Для дальшейших исследований будем смотреть на одно- и трехдоменные архитектуры, так как в ходе улучшения выравнивания их выборка оказалась наиболее удачной.
Закодировала доменные архитектуры (1 для MipZ (PF09140); 3 для DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609); 2 для MipZ (PF09140) + ParA (PF10609)), закодировала сравниваемые таксоны (B - Bacteria, A - Archea, E - Eukaryota). К именам всех последовательностей спереди добавила коды архитектуры и таксона. Построила филогенетическое дерево с помощью программы MEGA. Использовала методы Neighbor-Joining (Рис. 3 и 4) (не использует гипотезу о молекулярных часах и строит неукоренённое дерево) и UPGMA (Рис. 5 и 6) (предполагает молекулярные часы, строит укоренённое дерево) с параметрами по умолчанию.
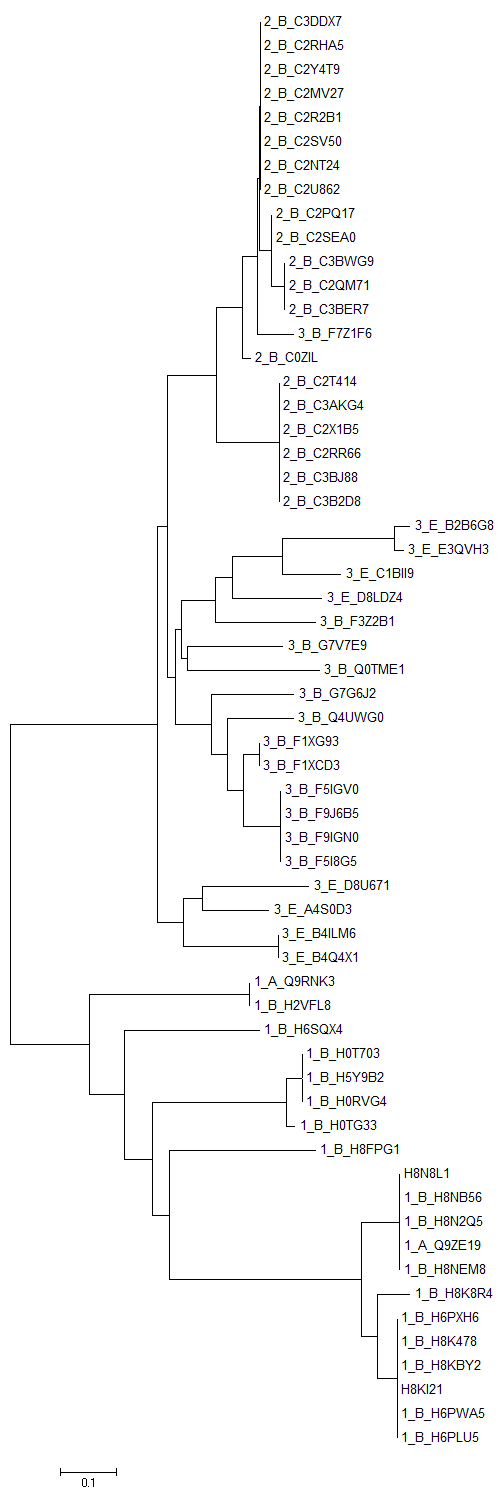
Рисунок 3. Филогенетическое дерево, построенное с использованием метода Neighbor-Joining.

Рисунок 4. Филогенетическое дерево, построенное с использованием метода Neighbor-Joining (топология).

Рисунок 5. Филогенетическое дерево, построенное с использованием метода UPGMA.

Рисунок 6. Филогенетическое дерево, построенное с использованием метода UPGMA (топология).
Создала рисунки деревьев с использованием методов выделения ветвей для наглядной демонстрации результата с помощью программы ITOL онлайн (рис. 7 и 8).
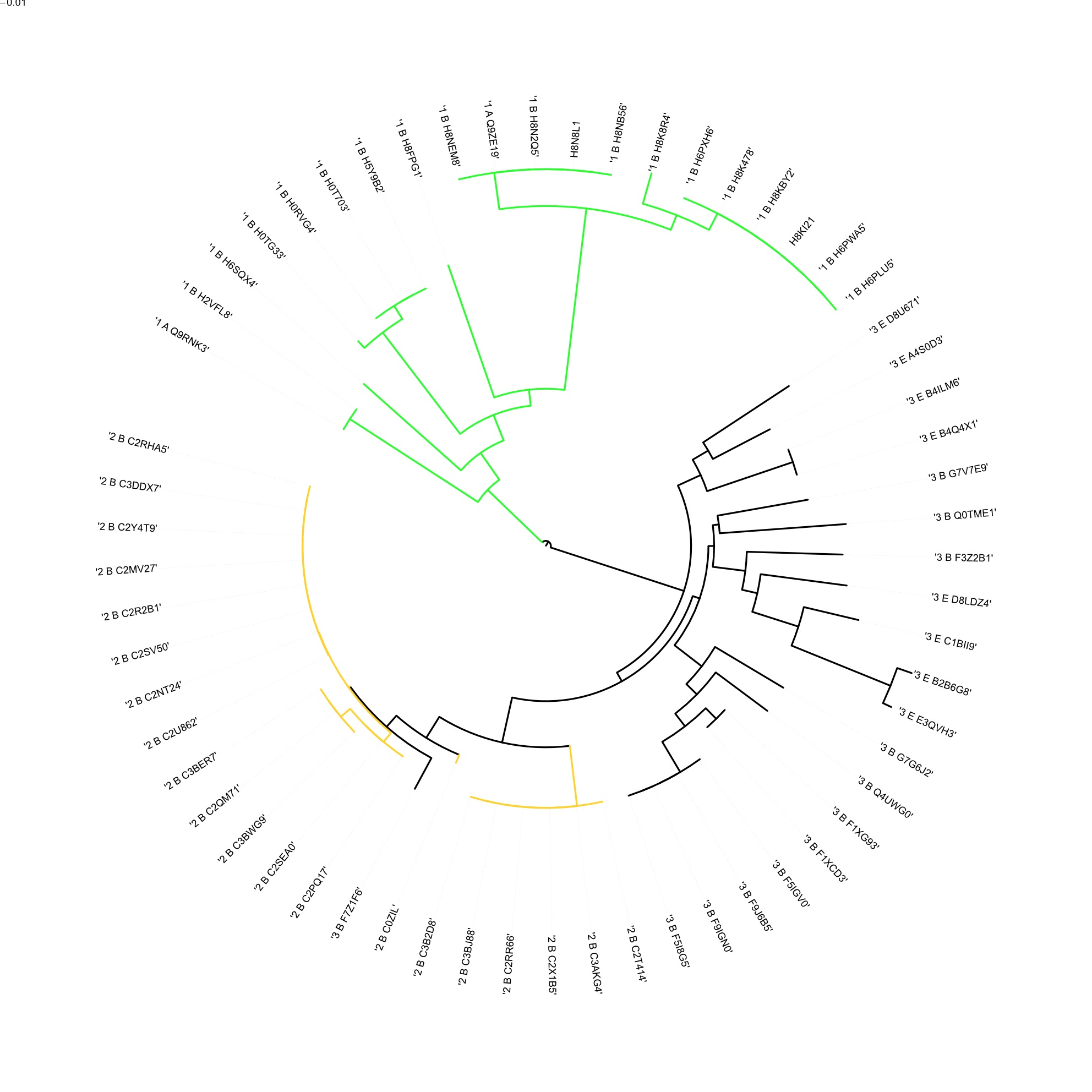
Рисунок 7. Дерево, построенное с помощью программы ITOL и метода Neighbor-Joining. Зеленым выделена ветвь однодоменного белка, желтым - однодоменного, черным - трехдоменные белки.
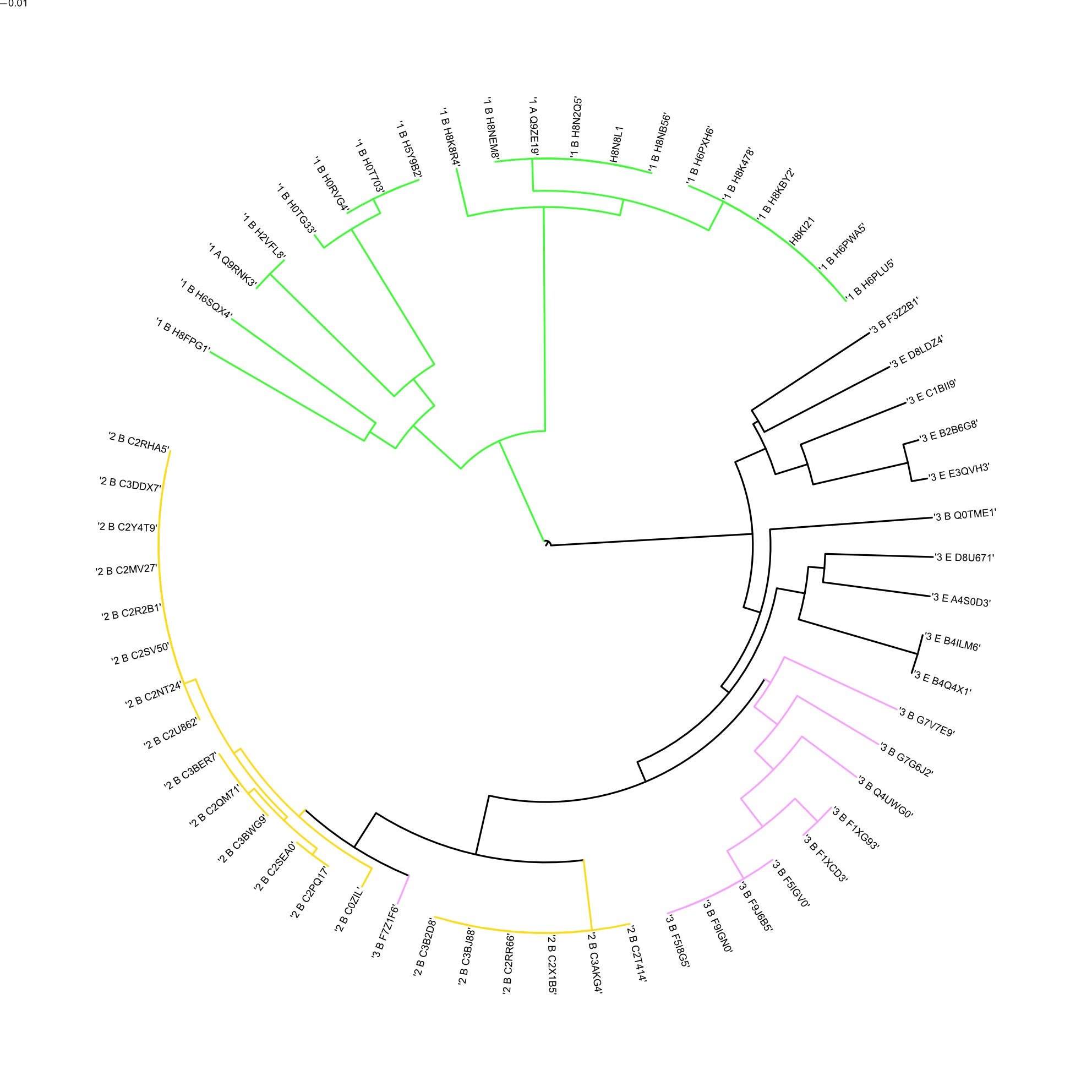
Рисунок 8. Дерево, построенное с помощью программы ITOL и метода UPGMA. Зеленым выделена ветвь однодоменного белка, желтым - однодоменного, черным - трехдоменные белки. Розовым показана выделившаяся ветвь трехдоменных белков у бактерий.
Скобочные формулы деревьев, построенных методом Neighbor-Joining (1.nwk) и UPGMA (2.nwk) в формате Newick с длиной ветвей.
На получившихся деревьях видно, что домены из разных доменных архитектур хорошо различаются программой. На получившихся деревьях видно, что однодоменные белки отделились раньше остальных доменов; двухдоменные белки выделились из трехдоменных, которые в свою очередь, имеют более разветвленные ветви, что означает высокую полиморфность этих белков.
Выделила из семейства Pfam подсемейство вида "домены семейства PF09140 из белков таксона B (Bacteria) с доменной архитектурой DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609)". На дереве моей выборки домены подсемейства образовывают отдельную кладу.
На основании моей выборки построила профиль, выделяющий данное подсемейство из всего семейства Pfam с помощью программы пакета HMMER 2.3.2 hmm2build. Выделила последовательности подсемейства из выравнивания моей выборки в отдельное выравнивание . Программой hmm2build построила профиль по этому выравниванию. Программой hmm2calibrate откалибровала профиль.
Для проверки профиля создала файл в fasta-формате со всеми белками Uniprot, включающими хоть один домен из моего Pfam-семейства. Программой hmm2search провела поиск откалиброванным профилем по всем белкам Uniprot, включающим хотя бы один домен моего семейства.
Были получены файлы с профилем НММ и с результатами поиска по профилю в множестве белков с PFAM,
для которых было предсказано наличие домена. Для большинства белков была установлена гомология
с доменом (E-value < 1*Е-5).
1. Выравнивание, по которому строился профиль
2. Построенный профиль
3. Выдача поиска
Далее была построена roc-кривая, изображенная на рисунке 9.
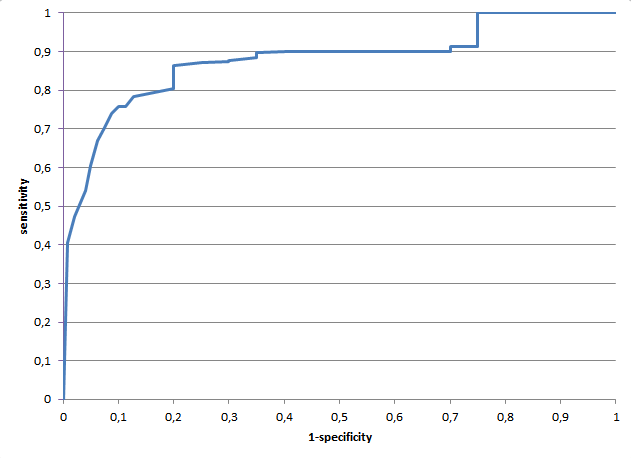
Рисунок 9. roc-кривая
Характеристики работы профиля по всем белкам Uniprot, включающим хоть один домен из моего Pfam-семейства(при пороге на E-value 0.00001):
Характеристики работы профиля с порогом e-value 1*E-5:
| Принадлежит подсемейству | Не принадлежит подсемейству | Всего | |
| Выше порога | 117 | 242 | 359 |
| Ниже порога | 0 | 60 | 60 |
| Всего | 117 | 302 | 419 |
чувствительность ("True positive rate") R = TP/(TP+FN) = 1;
избирательность ("Positive prediction value") PPV = TP/(TP+FP) = 0.326.
Получается, что при выбранном пороге Е-значения скорее будет дан ложноотрицательный результат, чем ложноположительный. Таким образом, данный профиль можно использовать только для грубой оценки.
Дата последнего обновления: 15.09.2014
Copyright © Кузнецова Мария, 2013.