Учебный сайт Кузнецовой Марии
Построение дерева по нуклеотидным последовательностям
Из базы полных геномов NCBI были получены последовательности 16S рибосомальной РНК каждой из бактерий (для каждой бактерии был
использован файл *.frn). Все последовательности были записаны в файл, в названиях последовательностей были оставлены только соответствующие мнемоники
бактерий.
Далее последовательности были выравнены с помощью сервера Muscle, выравнивание было импортировано в MEGA (при импорте был указан метод Analyze).
Дерево было реконструировано методом Maximum likelihood. Графическое изображение дерева можно увидеть на рис. 1.
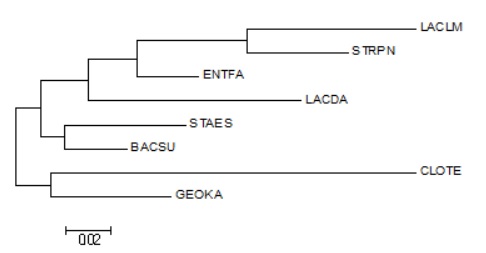
Рис. 1. Филогенетическое дерево нескольких видов бактерий отдела Firmicutes, построенное на основе выравнивания нуклеотидных последовательностей РНК малой субъединицы рибосомы
(16S rRNA). Дерево построено методом Maximum likelihood.
Если сравнить полученное дерево с правильным деревом, построенным на основе филогении (см. предыдущий практикум), то можно заметить, что правильно реконструированы только три
нетривиальные ветви - {STRPN, LACLM} vs {ENTFA, LACDA, STAES, BACSU, GEOKA, CLOTE}, {STRPN, LACLM, ENTFA} vs {LACDA, STAES, BACSU, GEOKA, CLOTE} и {STRPN, LACLM, ENTFA, LACDA} vs
{STAES, BACSU, GEOKA, CLOTE}. Остальные нетривиальные ветви правильного дерева отсутствуют на полученном. Дерево, построенное на основе последовательностей 16S РНК значительно
хуже отражает действительность, чем деревья, построенные на основе выравнивания белков (полученные во время выполнения предыдущих практикумов). Это связано с тем, что
выравнивания нуклеотидных последовательностей сами по себе менее достоверны, чем выравнивания соответствующих белков (из-за того что нуклеотидные последовательности состоят
только из 4-ёх букв, вероятность случайного совпадения позиций намного больше, чем для белковых последовательностей).
Построение и анализ дерева, содержащего паралоги
Проводился поиск гомологов белка CLPX_BACSU среди белков восьми выбранных бактерий.
Протеомы бактерий были скачаны из директории P:\y13\term4\Proteomes и объединены в общий файл all_my_proteomes.fasta. Файл с
последовательностью исследуемого белка был скачан из базы данных Swissprot.
Поиск гомологов проводился с помощью blastp:
makeblastdb -in all_my_proteomes.fasta -dbtype prot
blastp -query CLPX_BACSU.fasta -db all_my_proteomes.fasta -evalue 0.001 -outfmt 7 -out blastp.out
На основе полученного файла, содержащего информацию о гомологах, был создан файл-список:
cat blastp.out | egrep -v '#' | awk ' {print $2}' | awk ' BEGIN {FS = "|"} {print "fasta::all_my_proteomes.fasta:" $3}' | sort -u > homolog_id.bs
Получившийся список homolog_id.bs был подан на вход программе seqret:
seqret @homol_un_id.bs
Таким образом был получен файл homolog_seq.fasta, содержащий аминокислотные последовательности гомологов белка CLPX_BACSU. В названиях последовательностей были оставлены
только идентификаторы белков, после чего последовательности были выравнены с помощью Muscle, выравнивание импортировано в MEGA.
Дерево реконструировано методом Maximum likelihood. Графическое изображение получившегося дерева можно увидеть на рис. 2.
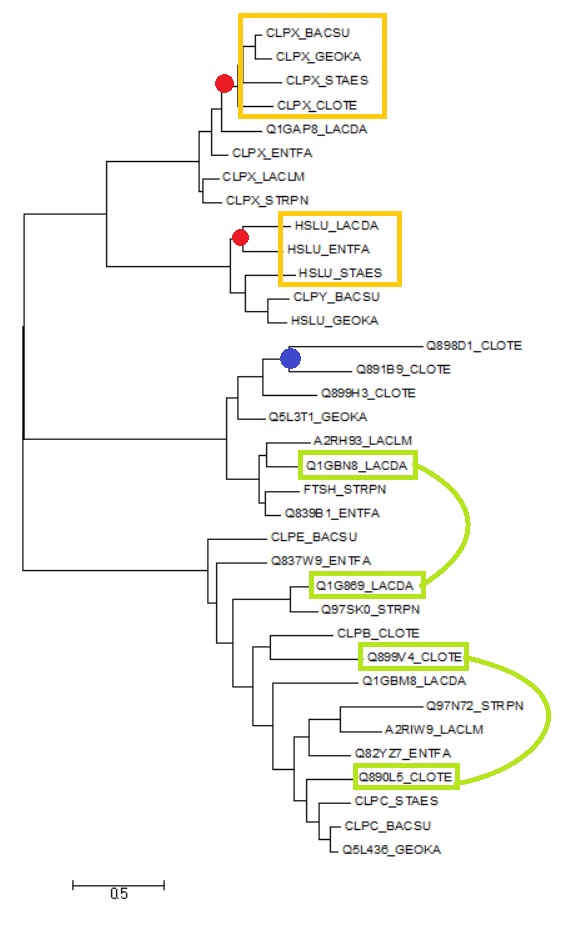
Рис. 2. Филогенетическое дерево гомологов белка CLPX_BACSU среди белков восьми бактерий. Построено методом Maximum likelihood. Желтыми рамочками выделены группы попарно ортологичных белков, зелеными - паралоги. Красными кружочками помечены примеры разделения путей эволюции белков в результате видообразования, синим кружочком - пример дупликации гена.
Дата последнего обновления: 15.09.2014
Copyright © Кузнецова Мария, 2013.