Ресеквенирование. Поиск полиморфизмов у человека.
1. Описание полиморфизмов пациента. Подготовка чтений.
В качестве исходных данных были использованы чтения экзома, картирующие участок 13-й хромосомы человека.
Файл с одноконцевыми чтениями в формате fastq: chr13.fastq. Хромосома человеческого генома (сборка версии hg19): chr13.fasta.
В директории morozova_ea@kodomo.fbb.msu.ru:/nfs/srv/databases/ngs/morozova_ea/ была создана моя директория morozova_ea, куда были скопированы файлы с ридами (chr13.fastq) и хромосомой (chr13.fasta).
1. Анализ качества чтений
Далее, с помощью программы FastQC был осуществлен контроль качества чтений: fastqc chr13.fastq
В результате был получен архив chr13_fastqc.zip, содержащий отчет о работе программы в виде html-страницы chr13_fastqc.html
2. Очистка чтений
Для лучшего качества чтений необходимо провести очистку.Применим программу Trimmomatic. С конца каждого чтения будут отрезаны нуклеотиды с качеством не ниже 20 (TRAILING:20) и оставлены только чтения длиной не меньше 50 нуклеотидов (MINLEN:50):
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq trimm_out.fastq MINLEN:50 TRAILING:20
В результате получен файл trimm_out.fastq.
Очищенная последовательность была также проанализирована программой FastQC: fastqc trimm_out.fastq и получена страница trimm_out_fastqc.html.
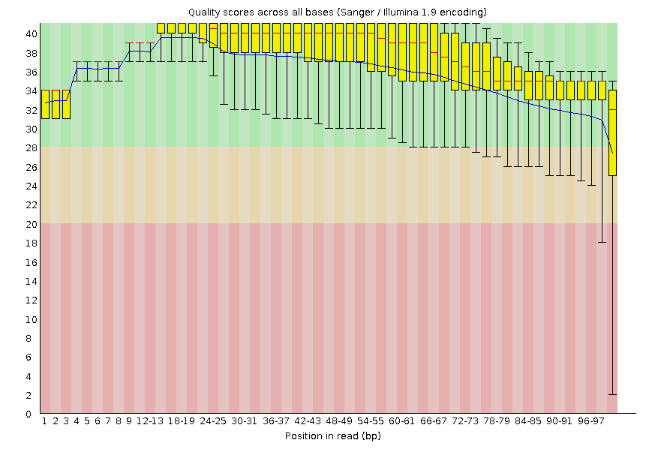
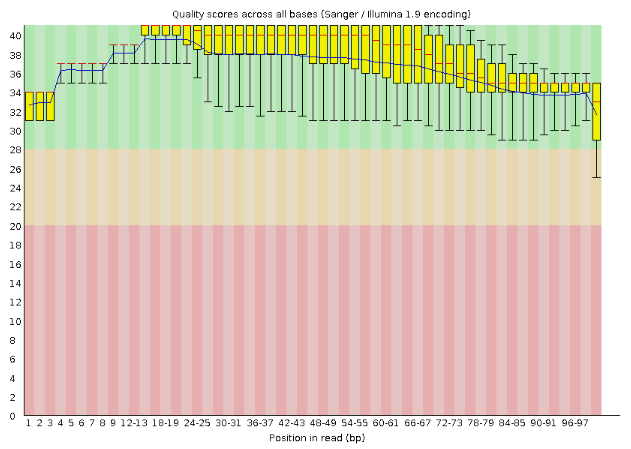
На рисунках 1(до очистки) и 2 (после очистки)наглядно показаны результаты работы программы FastQC, то есть качество прочтений до и после очистки по всем основаниям в каждой позиции fastq-файла.
Рис.1
Рис.2
Для каждой позиции построена диаграмма размахов.
Желтые прямоугольники - это интерквартильные размахи (от 25 о 75%), синяя линия - математическое ожидание, концы черных линий - точки от 10 до 90%, красная линия - медиана.
Чем больше значение по оси Y, тем лучше определено основание. По цвету фона также можно судить о точности: зеленая область фона - качество хорошее, оранжевая - среднее, красная - плохое.
После очистки число ридов уменьшилось на 6 (с 12155 до 12149), так как были удалены риды наиболее низкого качества.
Видно, что после очистки осле все чтения оказались в "зеленой области". То есть, благодаря работе программы Trimmomatic, были получены приемлимые для дальнейшей работы чтения.
3. Картирование чтений
Полученные чтения необходимо откартировать с помощью программы BWA (Burrows-Wheeler Alignment Tool).
На первом этапе хромосома 13 была проиндексирована:
bwa index chr13.fasta
Затем было построено выравнивание прочтений и проиндексированной референсной последовательности:
bwa mem chr13.fasta trimm_out.fastq >> align_pr13.sam
Получен файл align_pr13.sam с выравниваниями в формате sam.
4. Анализ выравнивания
Полученное выравнивание чтений с референсом было переведено в бинарный формат bam:
samtools view -b align_pr13.sam -o align_pr13.bam
Параметр -b обозначает, что формат выходного файла - bam, параметр -o обозначает выходной файл (align_pr13.bam).
Далее выравнивание чтений с референсом (получившийся после картирования align_sort.bam файл) было отсортировано по координате в референсе начала чтения:samtools sort align_pr13.bam align_sort
Cортированный файл был проиндексирован:samtools index align_sort.bam
Было выяснено, сколько чтений откартировалось на геном:samtools idxstats align_sort.bam >> number.out
В файле number.out содержится информация, что из 12149 чтений на хромосому были откартированы 12051.5. Поиск SNP и инделей
Для начала был создан файл с полиморфизмами в формате .bcf:
samtools mpileup -uf chr13.fasta align_sort.bam -o snp.bcf
Затем, был создан vcf-файл со списком отличий между референсом и чтениями: bcftools call -cv snp.bcf -o snp.vcf.
Всего в файле snp.vcf указано 135 полиморфизма, из них 6 вставок, 7 делеций и 122 замены.
Среднее значение качества - 79.2, покрытия - 33.7. Полученные результаты можно считать приемлимыми.
В таблице №1 описаны в качстве примера 3 полиморфизма, найденные в файле snp.vcf.
Информация о полиморфизмах 6. Аннотация SNP
С помощью программы annovar необходимо проаннотировать только snp (без инделей), используя базы данных: refgene, dbsnp, 1000 genomes, GWAS и Clinvar.
Сначала необходимо подготовить входной файл для корректной работы программы.
В первую очередь из файла snp.vcf вручную были удалены индели (получили файл snp_only.vcf).
Дальнейшая подготовка файла:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput
Затем, полученный файл snp.avinput использовался для аннотации SNP по базам данных, указанным выше, с помощью скрипта annotate_variation.pl.6.1 Аннотация по базе refgene
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr13.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
База данных refseq в annovar делит SNP на несколько групп, которые отображены в первой колонке файла chr13.refgene.variant_function.
Эти категории отражают, где находится данный полиморфизм: внутри экзона, интрона, на пересечении генов, входит в 3'-нетранслируемую область, в 5'-нетранслируемую область, в пределах n bp от границы сплайсинга и т.д.
Получены файлы chr13.refgene.variant_function (описание всех SNP), chr13.refgene.exonic_variant_function (описание SNP, попавших в экзоны) и chr13.refgene.log (описание процесса работы программы).
В таблице №2 указана информация о SNP, принадлежащих различным группам, полученная из файла chr13.refgene.variant_function.
В таблице №3 представлена информация о SNP в экзонах, полученная из файла chr13.refgene.exonic_variant_function.
Всего в экзоны попало 7 SNP.
Данные о SNP в различных группах Таблица №2.
Данные о SNP в экзонах Таблица №3.
Как видно из таблиц, большинство замен произошли в интронах.
Возможно, это связано с тем, что замены в последовательностях кодирующих областей могут привести к изменению аминокислоты (несинонимичная замена) в конечном продукте - белке, чем нарушить его структуру и тем самым функцию.6.2 Аннотация по базе dbsnp
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Получены файлы chr13.dbsnp.hg19_snp138_dropped (содержит список SNP, имеющих rs, то есть аннотированные в этой базе данных),
chr13.dbsnp.hg19_snp138_filtered (SNP не аннотированные в базе данных) и chr13.dbsnp.log (описание процесса работы программы).
Выяснилось, что 109 SNP имеют аннотацию в базе данных dbSNP, а 13 не имеют.6.3 Аннотация по базе 1000 genomes
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Получены следующие три файла: chr13.1000.hg19_ALL.sites.2014_10_dropped, chr13.1000.hg19_ALL.sites.2014_10_filtered, chr13.1000.log, аналогичные выходным файлам в предыдущем пункте.
105 имеют аннотация, а 17 не имеют.
Также были выяснены частоты аннотированных полиморфизмов. Она варьируется от 3,55 до 225,01. В среднем частота составляет 87,59.6.4 Аннотация по базе GWAS
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr13.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Получено 2 файла: chr13.gwas.hg19_gwasCatalog (SNP, для которых известно клиническое значение) и chr13.gwas.log (описание процесса работы программы).
Среди исследуемых полиморфизмов 3 имеют аннотированное клиническое значение. Один связан с артериальной жесткостью, другой - гипертонией, третий связан с ожирением.6.5 Аннотация по базе Clinvar
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
Получено 3 файла chr13.clincar.hg19_clinvar_20150629_dropped, chr13.clincar.hg19_clinvar_20150629_filtered, chr13.clincar.log.
Оказалось, что в этой базе данных ни один из изучаемых SNP не аннотирован.
Итоговый excel-файл по всем SNP: pr12.xlsx.