Поиск сигналов. Chip-seq.
1. Контроль качества прочтений.
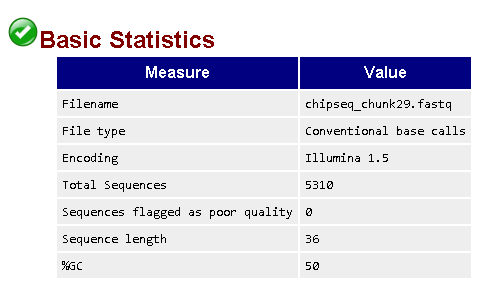
С помощью программы FastQC была произведена оценка качества ридов (файл с ридами chipseq_y14/chipseq_chunk29.fastq, который был предварительно скопирован из папки /P/srv/databases/ngs/chipseq_y14 в рабочую директорию /nfs/srv/databases/ngs/morozova_ea/chipseq_chunk29.fastq).
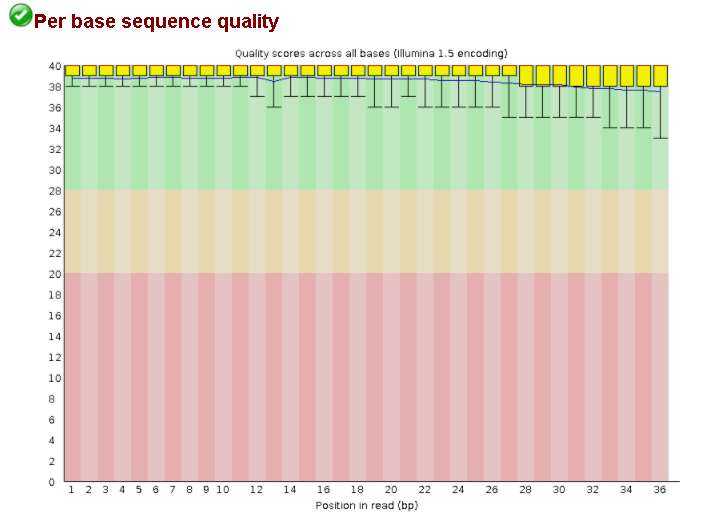
fastqc chipseq_chunk29.fastqВ результате был получен файл с расширением html. Количество чтений составило: 5310. Согласно полученной сводке мой файл не содержал последовательностей низкого качества. Распределение per base quality также оказалось хорошего качества. Как видно на рис.2 концы усов на диаграмме лежат в области приемлемого высокого качества. В этой связи дополнительная чистка чтений (с помощью Trimmomatic) не нужна.
Рис.1.
Рис.2. Качество по нуклеотидам из выдачи FastQC
2. Картирование прочтений на геном человека.Далее, с помощью алгоритма BWA было необходимо откартировать прочтения на геном человека h19 (файл из директории /srv/databases/ngs/hg19). На выходе получаем выравнивание в формате sam.
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk29.fastq > chipseq_chunk29.samДля анализа выравнивания файл chipseq_chunk29.sam был переведен в бинарный формат bam с помощью пакета samtools
samtools view chipseq_chunk29.sam -b -o chipseq29.bamДалее провели сортировку bam-выравнивания по координате начала прочтения в референсе. Параметр -T нужен для указания пути, по которому сохраняются временные файлы. В итоге имеем файл sorted.chipseq29.bam.
samtools sort chipseq29.bam -T chip -o sorted.chipseq29.bamИндекcирование отсортированного файла:
samtools index sorted.chipseq29.bamЗатем полученный файл был исследован на предмет числа откартированных чтений на геном с помощью команды
samtools idxstats sorted.chipseq29.bam > 29.txtВ полученном файле 29.txt представлены картированные и некартированные чтения на хромосому соответственно: 4990 и 320. Больше всего чтений пришлось на хромосому 19, которые в дальнейшем будут использоваться для анализа.
3. Поиск пиков.
Для поиска пиков (peak calling) воспользовались программой MACS.
macs2 callpeak -n chip19 -t sorted.chipseq29.bam --nomodelБез nomodel программа выдает, что пиков слишком мало.Опция -n служит для изменения названия эксперимента. Получены файлы, содержащие информацию о найденных пиках: chip19_peaks.narrowPeak, chip19_peaks.xls, chip19_summits.bed. Всего было найдено 5 пиков. Данные о них представлены в таблице ниже.
Номер пика
Координата начала
Координата конца
Длина
Расстояние вершины от старта пика
-Log10 (p-value)
-Log10 (q-value)
1
42772392
42772746
355
160
55.72848
46.25996
2
42786033
42786249
217
121
21.30536
14.35271
3
42901021
42901302
282
138
26.68766
19.53301
4
42924215
42924456
242
124
11.94254
5.27405
5
42954513
42954751
238
188
11.12271
4.49115
Ширина найденных 5-ти пиков варьировалась от 217 до 355. Стоит тметить, что чем меньше значение отрицательного десятичного логарифма p-value и q-value, тем достоверность пиков выше. Таким образов все находки оказались достаточно надежными. Среди них можно в особенности выделить пики 1 и 3:
-log10(p-value): 55.73 и -log10(q-value): 46.26
Наихудшие же показатель был у пика 5:
-log10(p-value): 11.12 и -log10(q-value): 4.49
Тем не менее, данный пик все равно можно смело причислить к достоверным.
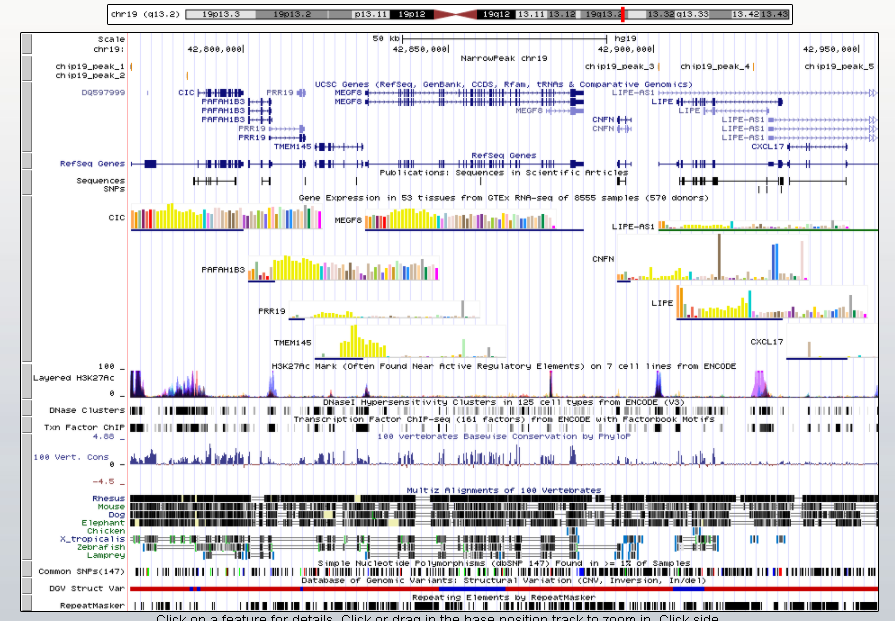
С помощью UCSC Genome Browser (My Data > Custom Tracks) была произведена визуализация пиков (рис. 3).
Рис.3. Визуализация пиков с помощью UCSC Genome Browser.
На вход подали: "track type=narrowPeak visibility=3 db=hg19 name="Peaks" description="NarrowPeak chr19" browser position chr19: 42772390-42954760" в начале файла chip19_peaks.narrowPeak.
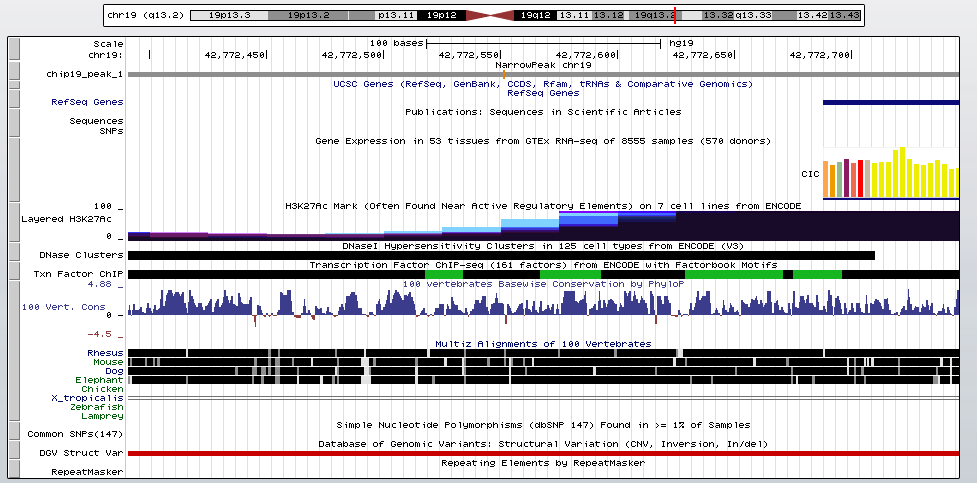
Для более детального рассмотрения был взят пик 1, как самый надежный. Ни с каким генами, однако, он не пересекается.
Рис.4. Пик 1.
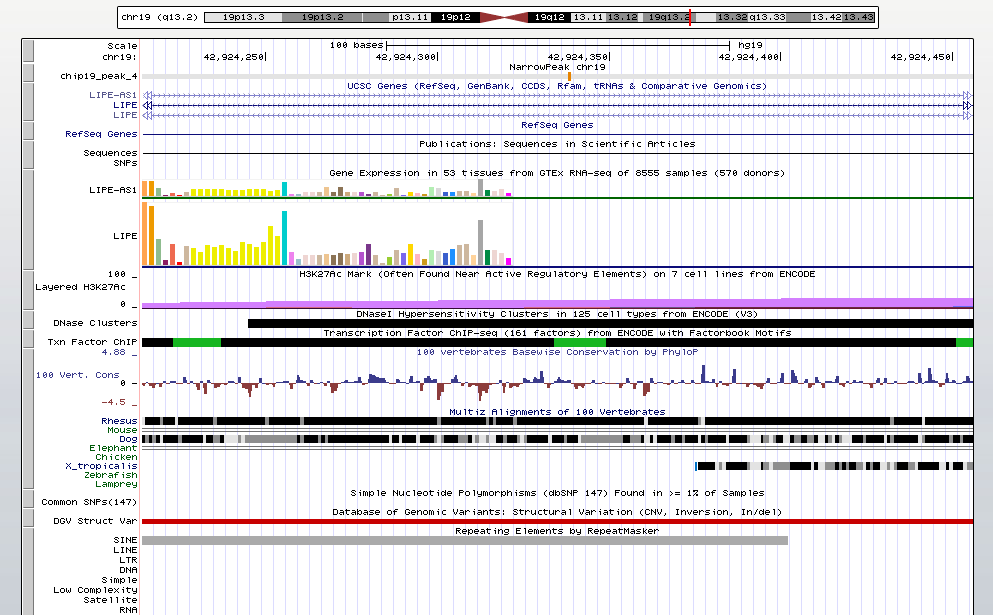
Также рассмотрели пик номер 4, который как видно попадает на интрон гена LIPE.
Рис.5. Пик 4.