Ресеквенирование. Поиск полиморфизмов
1.Номер хромосомы - 19
2.Таблица с использованными командами
| команда | функция |
| (1) fastqc chr19.fastq (2)fastqc outchr19.fastq |
анализ качества ридов до(1) и после(2) триммирования, на выходе архив с html-страничкой |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr19.fastq outchr19.fastq TRAILING:20 MINLEN:50 | очистка чтений;команда удаляет с конца каждого чтения нуклеотиды с качеством ниже 20 и оставляет только чтения длиной не меньше 50 нуклеотидов |
| hisat2-build chr19.fasta chr19 | индексирует референсную последовательность,выходные файлы chr19.?.ht2 |
| hisat2 -x chr19 -U outchr19.fastq --no-spliced-alignment --no-softclip -S chr19.sam | строит выравнивание прочтений и референса в формате sam, выходной файл - chr19.sam |
| samtools view -b -o chr19.bam chr19.sam | меняет формат .sam -> .bam |
| samtools sort chr19.bam chr19_sorted | сортирует выравнивание чтений с референсом по координате в референсе начала чтения, выходной файл - chr19_sorted.bam |
| samtools index chr19_sorted.bam | индексирует отсортированный .bam файл, выходной файл - chr19_sorted.bam.bai |
| samtools flagstat chr19.sam | статистика картирования ридов |
| samtools mpileup -uf chr19.fasta chr19_sorted.bam -o SNP.bcf | cоздаёт файл с полиморфизмами - SNP.bcf |
| bcftools call -cv SNP.bcf -o SNP.vcf | cоздаёт файл со списком отличий между референсом и чтениями - SNP.vcf |
| bcftools call -cvV indels SNP.bcf -o SNP_only.vcf | cоздаёт файл со списком отличий между референсом и чтениями - SNP_only.vcf (только SNP) |
| convert2annovar.pl --format vcf --outfile SNP_annot.avinput SNP_only.vcf | переводит .vcf в формат, доступный annovarу |
| annotate_variation.pl -out refgene_db_an -build hg19 SNP_annot.avinput /nfs/srv/databases/annovar/humandb | аннотация полиморфизмов по бд Refgene на основании генной разметки |
| annotate_variation.pl -filter -out dbsnp_fb_an -build hg19 -dbtype snp138 SNP_annot.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация полиморфизмов по бд SNP на основании фильтрации |
| annotate_variation.pl -regionanno -build hg19 -out gwas_rb_an -dbtype gwasCatalog SNP_annot.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация полиморфизмов по бд Gwas на основании разметки других регионов генома |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar_fb_an SNP_annot.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация полиморфизмов по бд Clinvar на основании фильтрации |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out genomes_fb_an SNP_annot.avinput /nfs/srv/databases/annovar/humandb.old/ | аннотация полиморфизмов по бд 1000 genomes на основании фильтрации |
3.Исходно получено 5524 чтения
4.Оценка качества чтений до триммирования
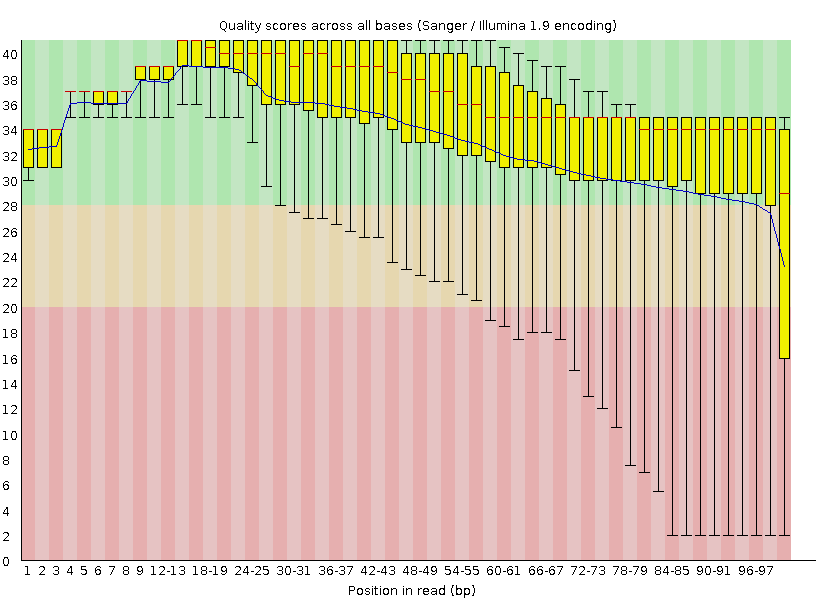
5.После триммирования осталось 5227 чтений.
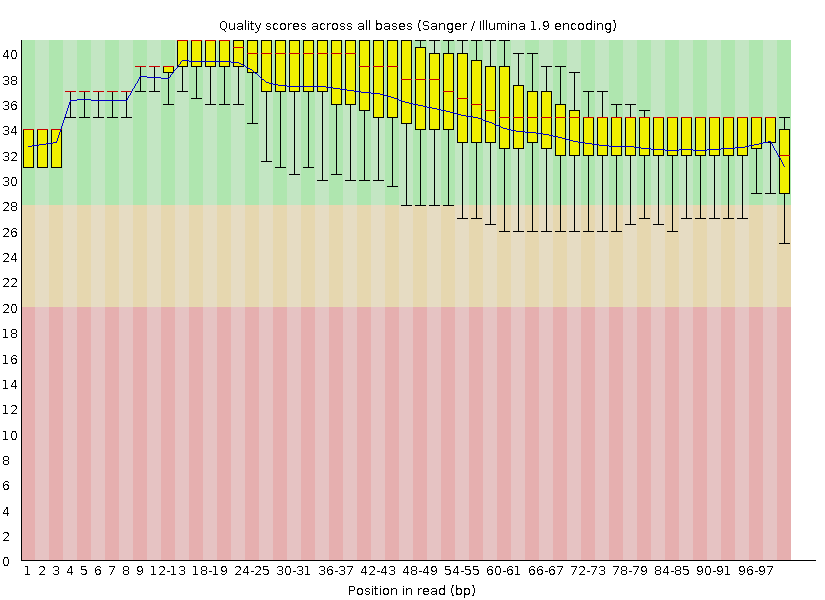
После триммирования общее качество ридов улучшилось, при этом их количество уменьшилось на 5,4%. Улучшение качества визуально заметно (рис.1 и рис.2), поэтому, на мой взгляд, триммирование в данных условиях оправданно.
6.На геном картировано 99.6% чтений.
С помощью программы samtools flagstat узнаём статистику:
5211 + 0 mapped (99.60%:-nan%)
7.Качество картирования очень хорошее, т.к. на геном ни разу не картировано 0.4 % ридов.
Вывод hisat2:
5227 reads; of these:
5227 (100.00%) were unpaired; of these:
21 (0.40%) aligned 0 times
5204 (99.56%) aligned exactly 1 time
2 (0.04%) aligned >1 times
99.60% overall alignment rate
8.Описание 3 полиморфизмов из .vcf файла
| координата (chr 19) | тип полиморфизма | референс | чтение | глубина покрытия | качество чтения |
| 17192567 | замена | С | T | 1 | 5.46383 |
| 17247445 | делеция | ca | c | - | 28.23 |
| 45406538 | вставка | CGGGGGGG | CGGGGGGGG | - | 24.5254 |
9. Было получено 87 SNP и 6 инделей.
| среднее покрытие | медианное покрытие | минимум покрытия | максимум покрытия | среднее качество | медианное качество | минимум качества | максимум качества |
| 13 | 8 | 1 | 124 | 83.555 | 54.0072 | 3.013 | 225.009 |
Поскольку качество > 20 считается "хорошим", то в данном случае качество полиморфизмов в среднем можно считать хорошим. Также видно, что риды картируются неравномерно, покрытие меньше 5 имеет примерно треть полиморфизмов, что наверное не очень хорошо.
10.Категории SNP базы данных RefSeq в annovar
| intronic | exonic | UTR3 | splicing | ncRNA | UTR5 | upstream | downstream | intergenic |
| 71 | 12 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
SNP попали в 3 гена: MYO9B, MPV17L2, TOMM40.
В экзонах имеется два типа нуклеотидных замены: nonsynonymous SNV (приводит к замене аминокислоты)-3 шт.
и synonymous SNV (не приводит к замене аминокислоты)-9 шт.
11. 77 SNP имеют rs.(получено из аннотации по базе SNP138)
12. 76 SNP проаннотировано по базе 1000Genomes, средняя частота SNP составляет 0.42
13. Клиническая аннотация: в бд Clinvar не нашлось ни одного из 87 SNP, по бд Gwas проаннотировано 4 полиморфизма:
| Name=Height | chr19 | 17283303 |
| Name=Multiple sclerosis | chr19 | 18304700 |
| Name=Alzheimer's disease (late onset),C-reactive protein,Alzheimer's disease biomarkers, AB1-42,Brain imaging,Longevity,Cholesterol, total,Cognitive decline,Cardiovascular disease risk factors, Alzheimer's disease,Age-related macular degeneration |
chr19 | 45395619 |
| Name=Metabolic syndrome | chr19 | 45396219 |