RNA-seq
1.Номер хромосомы - 19
2.Таблица с использованными командами
| команда | функция |
| (1) fastqc chr19.1.fastq (2)fastqc outchr19.1.fastq |
анализ качества ридов до(1) и после(2) триммирования, на выходе архив с html-страничкой |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr19.fastq outchr19.fastq TRAILING:20 MINLEN:50 | очистка чтений;команда удаляет с конца каждого чтения нуклеотиды с качеством ниже 20 и оставляет только чтения длиной не меньше 50 нуклеотидов |
| hisat2-build chr19.fasta chr19 | индексирует референсную последовательность,выходные файлы chr19.?.ht2 |
| hisat2 -x chr19 -U outchr19.1.fastq --no-softclip -S chr19.sam | строит выравнивание прочтений и референса в формате sam, выходной файл - chr19.1.sam; запускаем команду без параметра --no-spliced-alignment, т.к. он не разрешает картировать рид с разрывами, а в случае РНК-секвенирования это разрешение необходимо, т.к. могла быть прочитана зрелая мРНК, в которой отсутствуют интроны. |
| samtools view -b -o chr19.1.bam chr19.1.sam | меняет формат .sam -> .bam |
| samtools sort chr19.1.bam chr19.1_sorted | сортирует выравнивание чтений с референсом по координате в референсе начала чтения, выходной файл - chr19.1_sorted.bam |
| samtools index chr19.1_sorted.bam | индексирует отсортированный .bam файл, выходной файл - chr19.1_sorted.bam.bai |
| htseq-count -f bam chr19.1_sorted.bam -s no -i gene_id -m union gencode.v19.chr_patch_hapl_scaff.annotation.gtf > counts.txt |
считает, сколько чтений картировались на каждый ген; параметр -f определяет формат файла с чтениями, выравненными относительно хромосомы; параметр -s no не учитывает цепь параметр -i определяет, какой GFF атрибут будет использован в качестве feature ID параметр -m определяет, какие наложения ридов можно считать наложением на ген: union- любое пересечение гена и рида intersection_strict - весь рид попадает на ген(можно с разрывами) intersection_nonempty - любое пересечение гена и рида(но при пересечении с двумя генами, учитывается только один ген, с большим перекрытием) более подробно см картинку ниже. |
3.Исходно получено 34936 чтения
4.Оценка качества чтений до триммирования
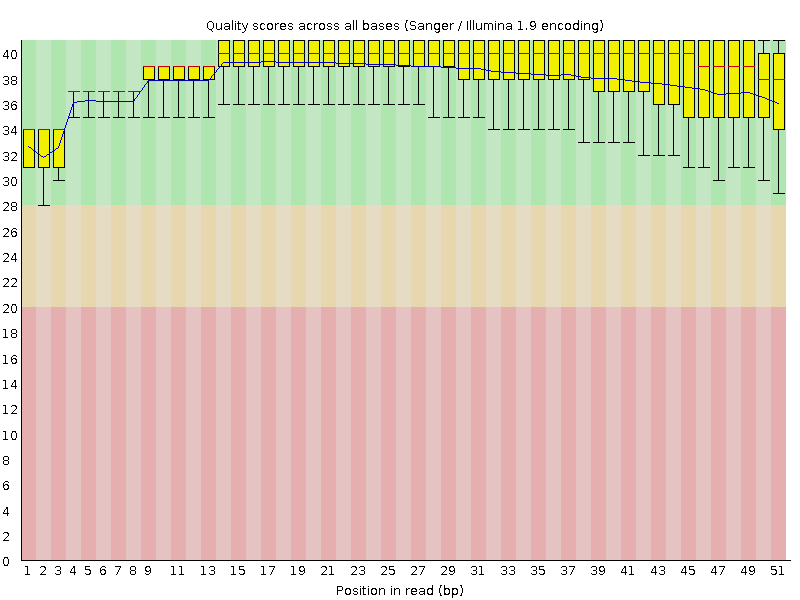
5.После триммирования осталось 34268 чтения.
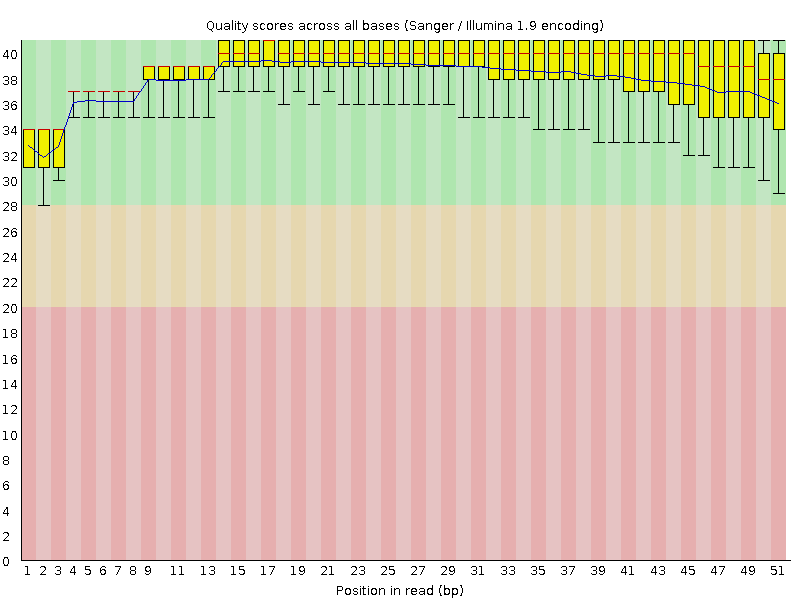
Результат работы trimmomatic:
Input Reads: 34936 Surviving: 34268 (98,09%) Dropped: 668 (1,91%)
Качество ридов улучшилось, но визуально этого практически не видно.
6.На геном картировано 98.52% чтений.
Качество картирования очень хорошее, т.к. на геном ни разу не картировано 1.48 % ридов.
Вывод hisat2:
34268 reads; of these:
34268 (100.00%) were unpaired; of these:
508 (1.48%) aligned 0 times
33760 (98.52%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.52% overall alignment rate
7.Htseq-count, параметр -m:
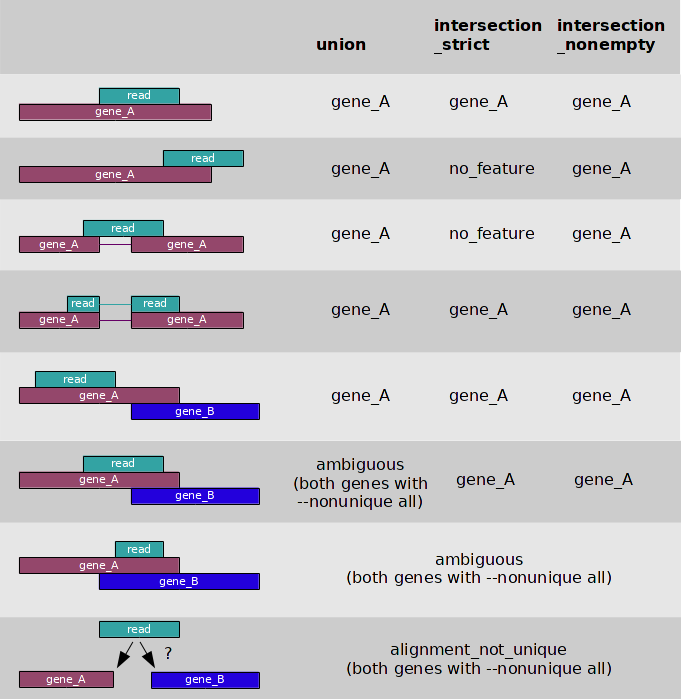
8. Результаты работы htseq-count (удалены все гены, перекрывающиеся с ридами 0 раз):
ENSG00000167658.11 33466
ENSG00000206775.1 6
__no_feature 281
__ambiguous 7
__not_aligned 508
Не все чтения легли в границы генов(281 из картированных не легли),скорее всего они попали в интроны. 7 чтений попали в оба гена.
ENSG00000167658 кодирует фактор элонгации трансляции 2 эукариот.
ENSG00000206775 кодирует малую ядрышковую РНК, которая участвует в метилировании 2'-O рРНК.