Задание 1. Предсказание вторичной структуры 1h4s-тРНК
Упражнение 1.
Нужно было найти инвертированные участки в заданной тРНК с помощью программы einverted. Если запускать программу с параметрами по умолчанию,
то количество находок комплементарных участков равно нулю. При уменьшении минимального количества очков (Minimum score threshold) с 50 до 10,
находится один комплиментарный участок длинной 7 п.о., при дальнейшем уменьшении данного параметра результат не изменяется (всё так же одна находка).
Варьирование других параметров либо даёт предыдущий результат (например, при изменении штрафа за gap), либо выдаёт слишком крупные участки.
Самый правдоподобный результат работы программы:
SEQUENCE: Score 21: 7/7 (100%) matches, 0 gaps
1 cggggag 7
|||||||
73 gcccctc 67
Упражнение 2.
Теперь попробуем найти инвертированные участки с помощью программы RNAfold. В этом случае полученный результаты были правдоподобнее, чем у einverted. Вот что получилось:
>1H4S:T|PDBID|CHAIN|SEQUENCE
CGGGGAGUAGCGCAGCCCGGUAGCGCACCUCGUUCGGGACGAGGGGGGCGCUGGUUCAGAUCCAGUCUCCCCGACCA
(((((((..((((.........)))).(((((((...))))))).....(((((.......)))))))))))).... (-33.80)
(((((((..{({{.,,..,,|.||}|.(((((((...})))))).}.)}||||{.......}}}}}))))))).... [-35.45]
(((((((....................(((((((...))))))).....(((((.......)))))))))))).... {-27.10 d=13.30}
(((((((..((((.........)))).(((((((...))))))).....(((((.......)))))))))))).... {-33.80 MEA=56.71}
frequency of mfe structure in ensemble 0.0686331; ensemble diversity 19.36
RNAfold предложила три варианта вторичной структуры заданной тРНК и выбрала наиболее стабильнуную (вторая структура, конечно, имеет более низкую энергию, но в отличие от первой содержит много пар со слабыми взаимодействиями между друг другом, и потому не выглядит многообещающей). Первая структура была визуализирована с помощью всё той же RNAfold (рис.1)
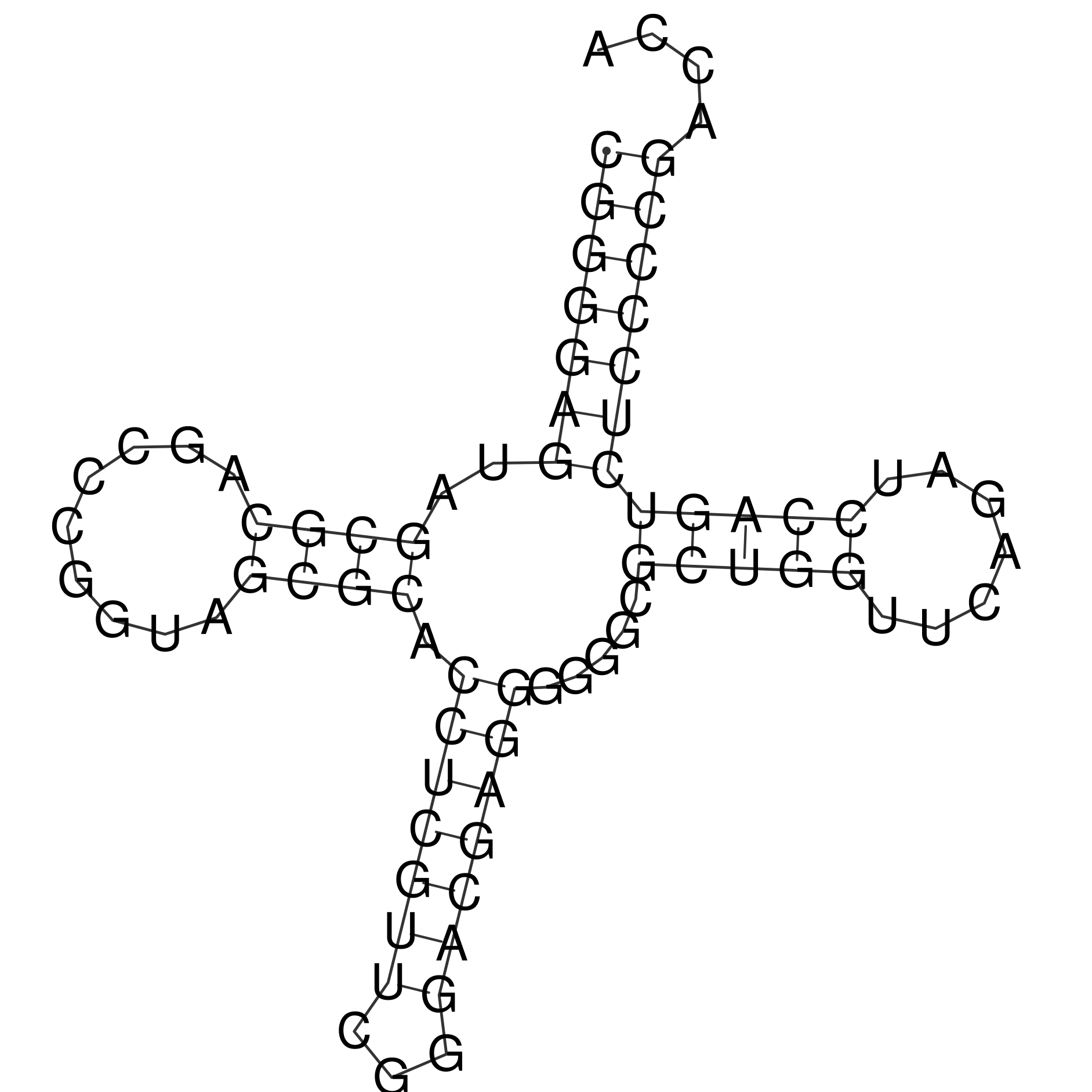
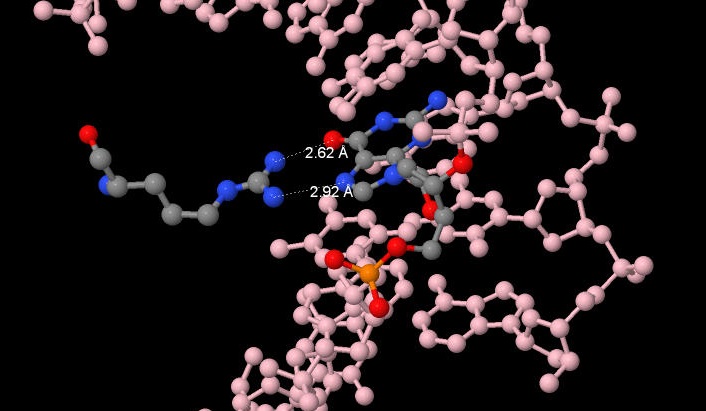
Как мы видим смоделированная структура действительно очень похоже на реальную, но вот антикодоновая петля, состоящая всего из 3 нуклеотидов вызывает большие сомнения.
Результаты сравнения реальной и предсказанных вторичных структур тРНК находятся в таблице 1.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 4-7 3' 5' 66-69 3' всего 4 пары |
предсказано 4 пары из 4 реальных и ещё 3 нереальных |
0 реальных пар 5' 1-7 3' 5' 67-73 3' |
| D-стебель | 5' 10-13 3' 5' 22-25 3' всего 4 пары |
- | 0 реальных пар 5' 10-13 3' 5' 23-26 3' |
| T-стебель | 5' 49-53 3' 5' 61-65 3' всего 5 пар |
- | 0 реальных пар 5' 50-54 3' 5' 62-66 3' |
| Антикодоновый стебель | 5' 26-32 3' 5' 38-44 3' всего 7 пар |
- | 0 реальных пар 5' 28-34 3' 5' 38-44 3' |
| Общее число канонических пар нуклеотидов | 18 пар | 4 пары | 21 пара |
Таким образом, структура, предсказанная по алгоритму Зукера, хоть и имеет низкую энергию и очень похожа на реальную структуру тРНК, но как мы видим из таблицы 1 не имеет ни одной верной пары (все пары смещены на 1-2 нуклеотида). Интересно, что данная тРНК имеет такую нуклеотидную последовательность, которая позволяет при смещении на 1-2 нуклеотида в сторону всё равно образовывать (в основном) канонические пары.
Задание 2.
Упражнение 1.
скрипт для define
скрипт, работающий в апплете
Упражнение 2.
скрипт
Поскольку в комплексе 1mhd есть 2 цепи белка, то при поиске контактов была выбрана только цепь А. Результаты поиска приведены в таблице 2.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 1 | 5 | 6 |
| остатками фосфорной кислоты | 5 | 1 | 6 |
| остатками азотистых оснований со стороны большой бороздки | 4 | 5 | 9 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
Общее количество ДНК-белковых контактов = 21.Наибольшее количество контактов у белка с остатками азотистых оснований со стороны большой бороздки, а вот с остатками азотистых оснований со стороны малой бороздки нет ни одного. Это кстати подтверждается с помощью визуализации комплекса в JMOL(рис.2, 3). Число белковых контактов с сахаро-фосфатным остовом и число белковых контактов с азотистыми основаниями примерно одинаково, что, на мой взгляд, свидетельствует о спецефичности взаимодействия в данном комплексе(ДНК-белок)
|
|
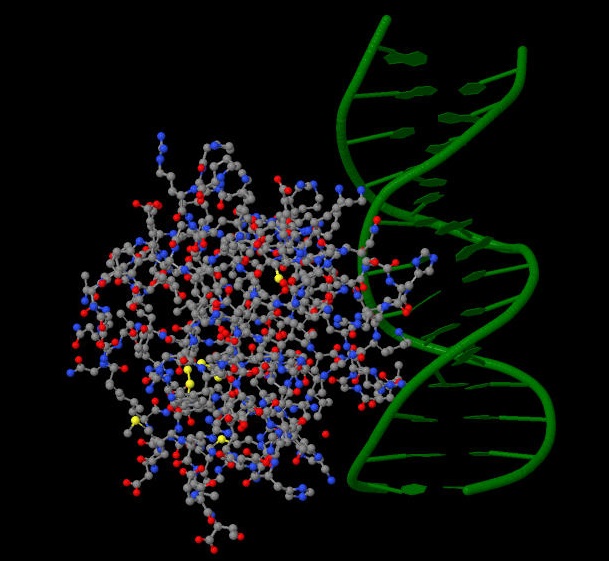
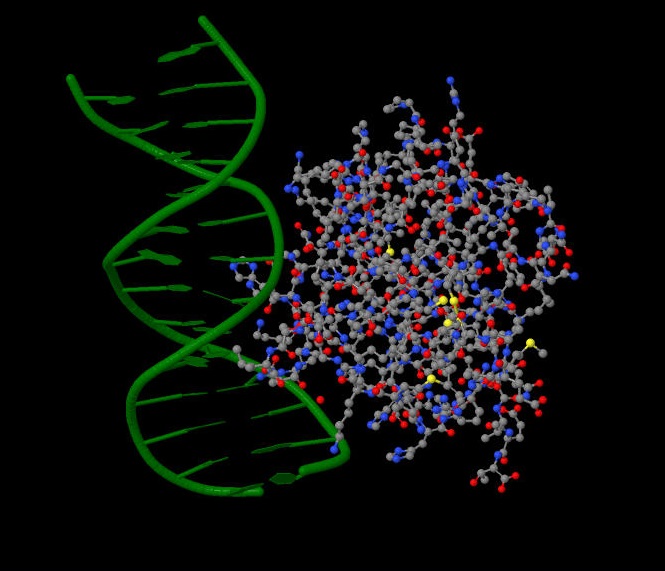
Упражнения 3,4.
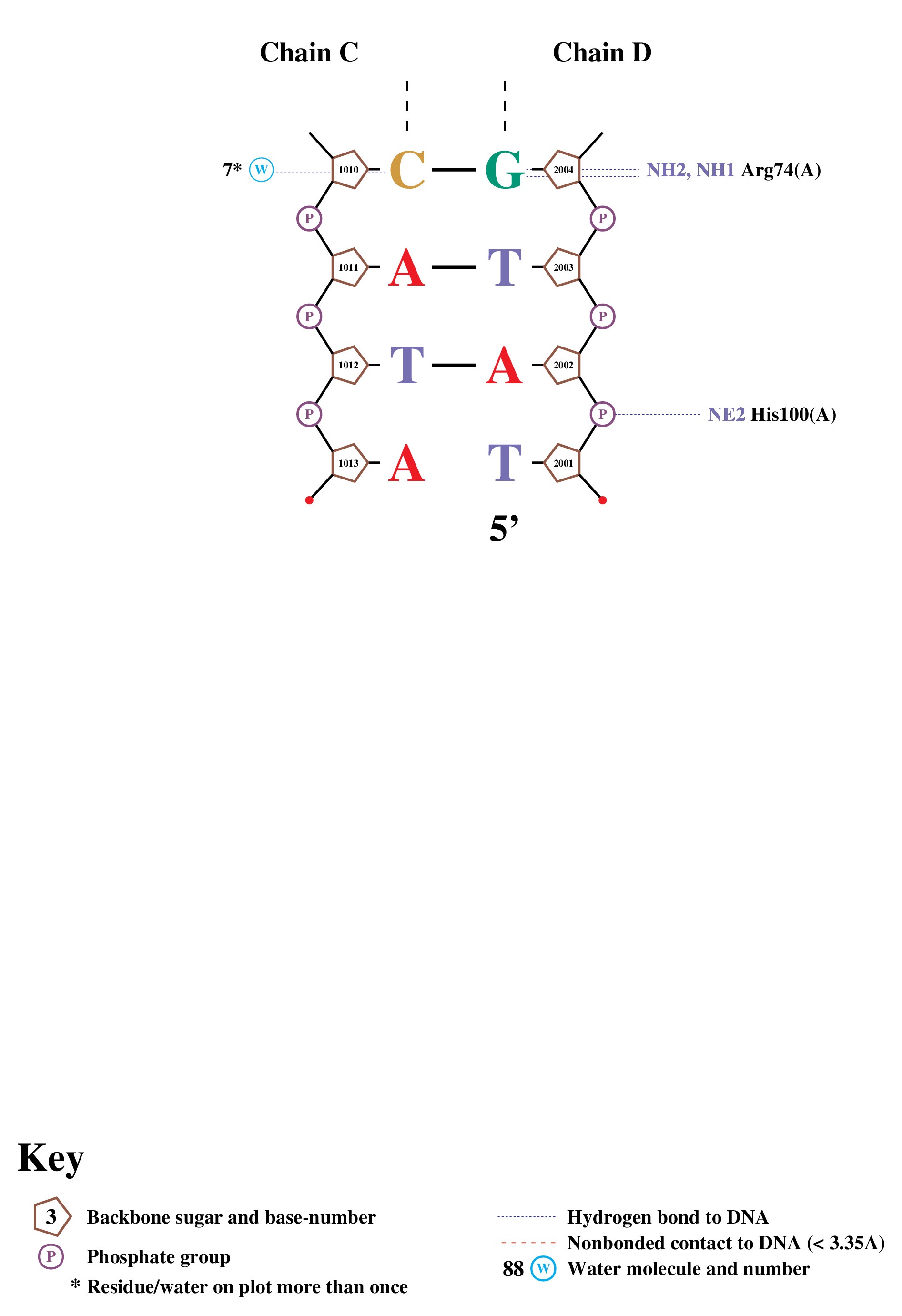
С помощью программы nucplot были получены схемы ДНК-белковых контактов(рис.4, 5)
|
|
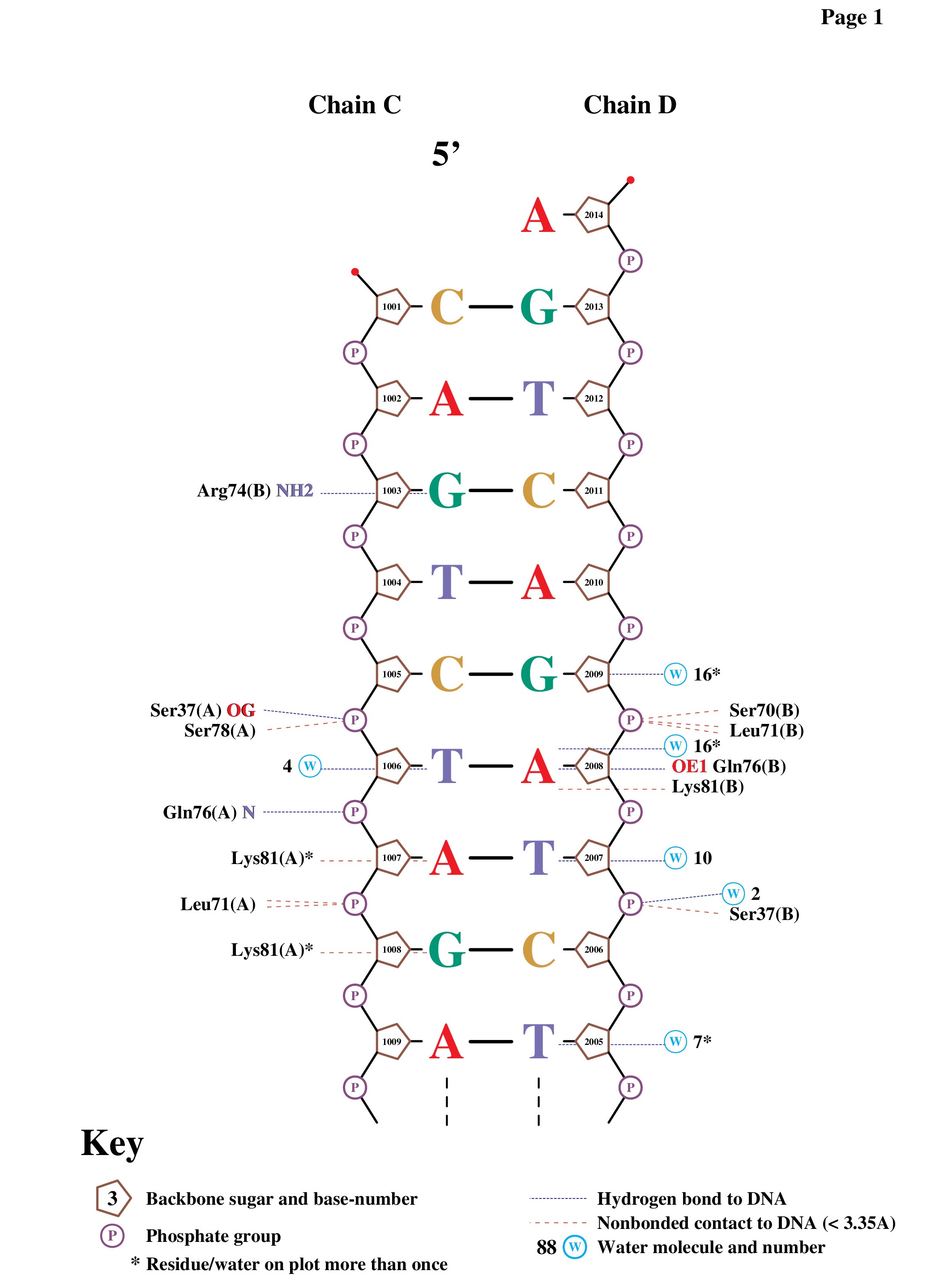
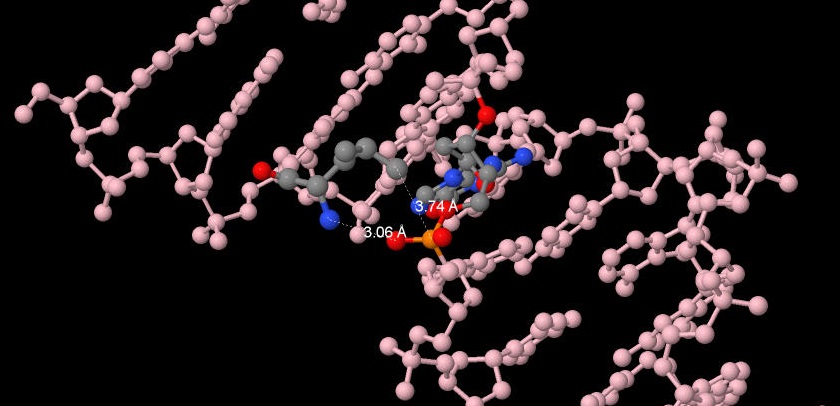
Leu(71:B) был выбран как "аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК"(рис. 6) Он имеет 1 полярный (между азотом и кислородом, S=3.06A) и 1 неполярный(между углеродом и фосфором, S=3.74А)
В качестве "аминокислотного остатка наиболее важного для распознавания последовательности ДНК" был выбран ARG(74:B)(рис.7), т.к. он образует две водородные связи непосредственно с азотистым основанием(G), а не с сахаро-фосфатным остовом.Это говорит о некоторой спецефичности данного взаимодействия.