1. Есть оптимизированная структура этана в виде z-matrix:
$DATA
eth
C1
C
C 1 cc
H 2 ch 1 cchv
H 2 ch 1 cch 3 d1 0
H 2 ch 1 cch 3 d2 0
H 1 ch 2 cch 3 d3 0
H 1 ch 2 cch 5 d3 0
H 1 chv 2 cch 4 d3 0
cc=1.52986
ch=1.08439
chv=1.08439
cch=111.200
cchv=111.200
d1=120
d2=-120
d3=180
$END
2. Создем файл-заготовку для размножения et.inp. Проверил работает ли он, для чего запускаес GAMESS и получаем выходной файл без ошибок.
3. Делаем скрипт make_b.bash для того чтобы создать 21 файл с разными длинами связей.
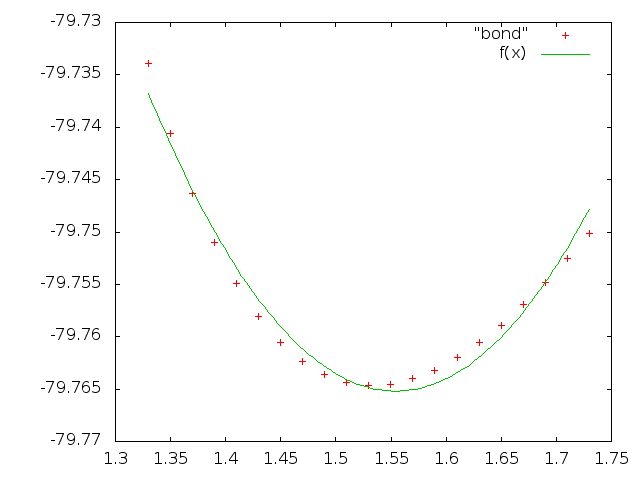
4. Добавляем в скрипт команды для запуска GAMESS и вывода нужных нам строк. В итоге получаем файл: bond.
5. Запускаем Gnuplot:
gnuplot
Сначала зададаем функцию в развернутом виде, в строке gnuplot введём:
f(x)=a + k*x*x - 2*k*x*b + k*b*b
И зададим стартовые значения коэффициентов:
a=-80
k=1
b=1.5
Проведём подгонку коэффициентов под имеющиеся точки в файле bond:
fit f(x) "bond" via a,k,b
Получаем значения коэффициентов:
a = -79.7652
k = 0.563608
b = 1.55432
Строим график:
set term 'png'
set output 'gnu_bond.png'
plot "bond", f(x)
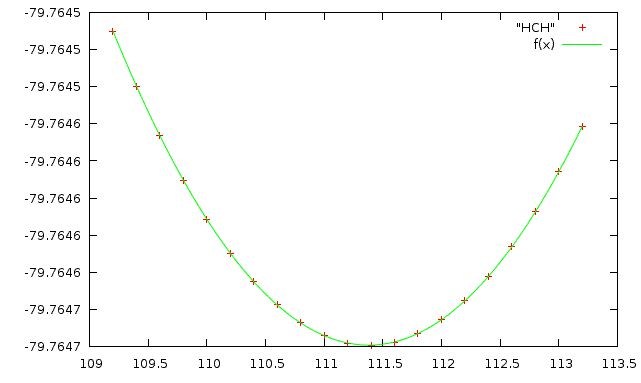
6. Сделаем аналогичные операции для валентного угла HCH
Его значения должны изменяться от 109.2 до 113.2
Скрипт для создания файлов: make_hch
Получаем файл: HCH
Получаем значения коэффициентов и график:
a=-79.7647
k=3.56075e-05
b=111.38
7. Сделаем аналогичные операции для для торсионного угла d3
Его значения должны изменяться от -180 до 180 c шагом 12
Получаем файл: torsion
Значения коэффициентов и график с апроксимированной зависимостью функицией:
f(x)=a*cos(k*x*pi/180)+b
a=0.00234519
k=3.00014
b=-79.7623
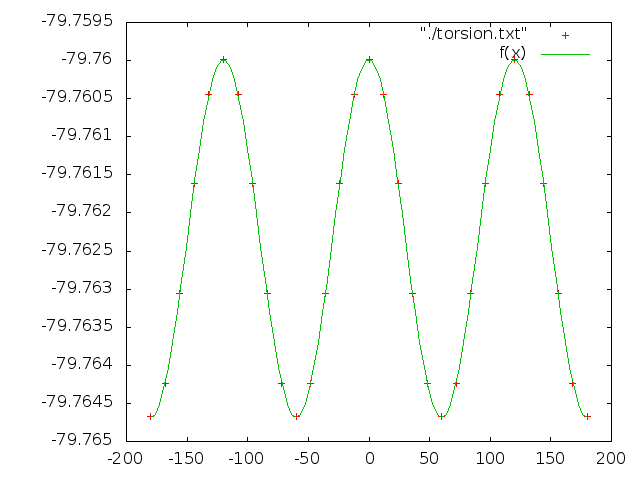
Так как крайние точки совпадают, получается 3 минимума.
