1)Сравнение программ MSA
Для сравнения использовалась программа Ксении Кирцовой.
Выравнивались последовательности 2m6b, 2lm2, 2lly(идентификаторы pdb). Доменная архитектура - один домен PF02932.
Сравнивались программы Clustal Omega, MAFFT, MUSCLE.
Ссылки на проекты с выравниваниями для разных программ:
Clustal_Omega MAFFT MUSCLEСравнение Clustal Omega и MAFFT:
Выравнивания достаточно сильно совпадают на участках 2-144 изначальных последовательностей, различия обнаруживаются ближе к C-концам. Выравнивание MAFFT содержит больше гэпов и инделей(вероятно, для нее штрафы за гэп и открытие инделя у нее несколько меньше, чем у Clustal W).
Выдача_сравнения_Clustal_Omega_и_MAFFTСравнение Clustal Omega и MUSCLE:
Данные выравнивания полностью совпали, что интересно и наводит на предположение, что это связано с эвристическими методиками и методом построения деревьев UPGMA, используемыми обеими этими программами, но не используемыми в MAFFT.
Выдача_сравнения_Clustal_Omega_и_MUSCLE2)Сравнение выравнивания тех же последовательностей с помощью Clustal Omega и их выравнивания по совмещению структур
Выравнивание по совмещению структур почти не совпало с выравниванием Clustal Omega. Наиболее сильно отличаются выравнивания для позиций у C-концов, которые в белках не входят в жесткую вторичную структуру. К тому же, по какой-то причине небольшое количество аминокислот их начала последовательностей, присутствующие в последовательностях из Uniprot, отсутствуют в выравнивании по совмещению структур(использовавшему записи из пдб). Судя по всему, это несколько сместило выравнивание и привело к такому огромному различию. Но так как эти аитнокислоты располагаются на N-концах, не вовлеченных ни в какую жесткую вторичную структуру, а вторичные структуры белков довольно сильно совпали, то можно предположить, что структурное выравнивание более правильное.
Проект Jalview с выравниванием по совмещению структур
Ссылка_на_выравнивание_на_сайте_PDB
Выдача_сравнения_Clustal_Omega_и_выравнивания_по_совмещению_структур
3)Описание программы MUSCLE
Программа множественного выравнивания MUSCLE(MUltiple Sequence Comparison by Log-Expectation) была создана Робертом Эдгаром в 2004 году. Общее описание алгоритма было опубликовано в Nucleic acid research(ссылка на статью приведена ниже).
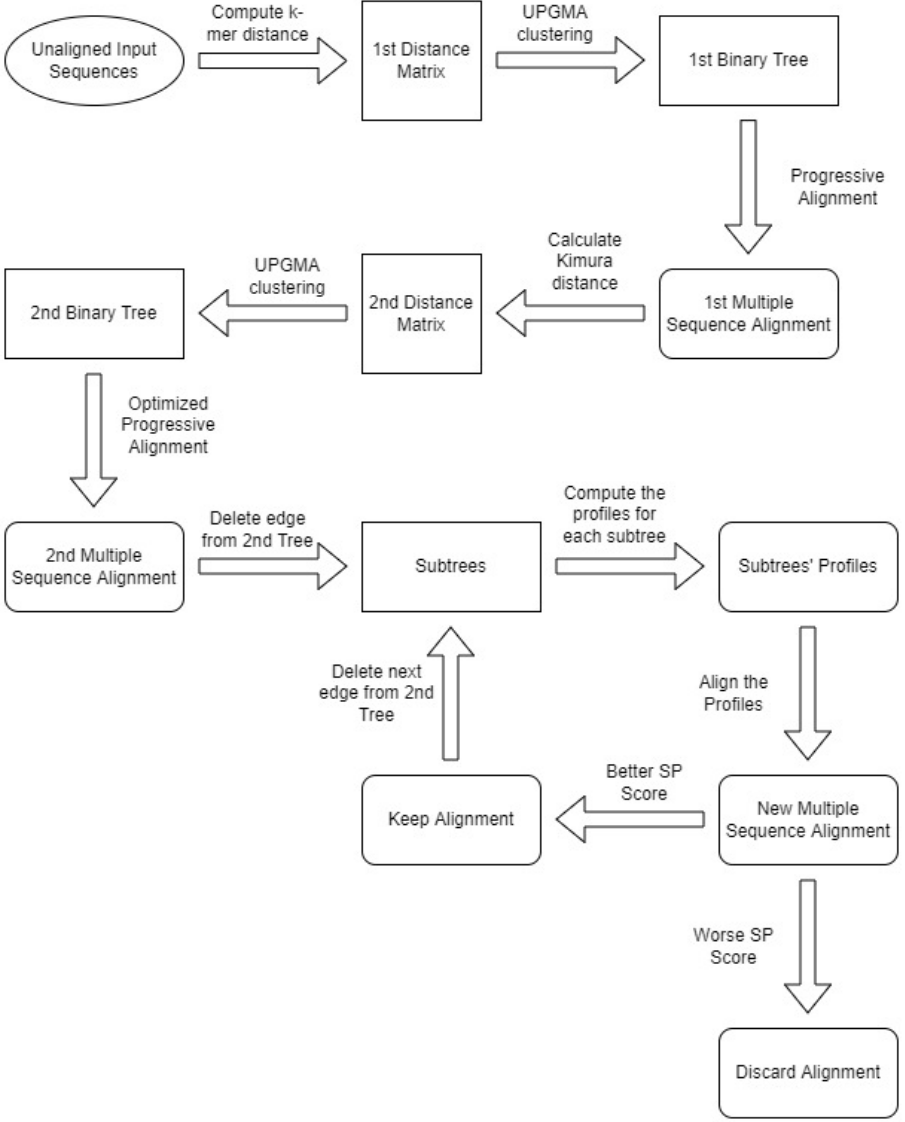
Если говорить о принципе работы алгоритма, то нужно будет упомянуть три стадии работы программы: черновое прогрессивное выравнивание, улучшенное прогрессивное выравнивание, рафинирование.
На стадии чернового выравнивания сравниваются k-меры(слова) из введенных последовательностей(последовательности длиной k), по ним строится матрица расстояний между последовательностями, которая далее преобразуется в дерево с помощью алгоритма UPGMA. Далее по полученному дереву строится прогрессивное выравнивание. Одно из главных преимуществ данного метода - высокая скорость работы первой стадии алгоритма, так как последняя не требует проведения выравниваний для построения дерева.
На стадии улучшенного выравнивания происходит первичное исправление некоторой неточности первой стадии. Расстояние между последовательностями рассчитывается по методу Кимуры, далее по этим данным алгоритмом UPGMA строится дерево, после чего для него создается новое прогрессивное выравнивание. При отличии структуры какого-либо участка второго дерева от первого дерева этот участок перевыравнивается в соответствии со вторым, в противном случае используется выравнивание, полученное на первой стадии.
На стадии рафинирования от листьев дерева к корню выбираются и удаляются ребра дерева. Таким образом одно дерево разбивается на два. Профили выравниваний двух отдельных деревьев выравниваются, при повышении счета оставляется новое выравнивание, в противном случае оно отбрасывется. Данный процесс продолжается до сближения результатов либо до установленного пользователем предела.
Ссылка_на_оригинальную_статью