Выделение доменов с помощью DOMAK¶
Реализуем алгоритм выделения структурных доменов DOMAK. Контакты между остатками посчитаем с помощью arpeggio.
необходимые импорты и установка arpeggio
!pip install -q condacolab
import condacolab
condacolab.install()
✨🍰✨ Everything looks OK!
!conda install -c conda-forge gemmi openbabel biopython
Channels:
- conda-forge
Platform: linux-64
Collecting package metadata (repodata.json): - \ | / - \ | / - \ | done
Solving environment: - \ | / done
==> WARNING: A newer version of conda exists. <==
current version: 23.11.0
latest version: 24.11.2
Please update conda by running
$ conda update -n base -c conda-forge conda
# All requested packages already installed.
!pip install pdbe-arpeggio
Collecting pdbe-arpeggio Downloading pdbe_arpeggio-1.4.4-py3-none-any.whl.metadata (9.7 kB) Requirement already satisfied: gemmi in /usr/local/lib/python3.10/site-packages (from pdbe-arpeggio) (0.7.0) Requirement already satisfied: numpy in /usr/local/lib/python3.10/site-packages (from pdbe-arpeggio) (2.2.1) Requirement already satisfied: biopython in /usr/local/lib/python3.10/site-packages (from pdbe-arpeggio) (1.84) Downloading pdbe_arpeggio-1.4.4-py3-none-any.whl (63 kB) ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 0.0/63.0 kB ? eta -:--:-- ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 63.0/63.0 kB 1.7 MB/s eta 0:00:00 Installing collected packages: pdbe-arpeggio Successfully installed pdbe-arpeggio-1.4.4
!pdbe-arpeggio -h
usage: pdbe-arpeggio [-h] [-s SELECTION [SELECTION ...] | -sf SELECTION_FILE] [-wh] [-mh]
[-ms MINIMISATION_STEPS] [-mf {MMFF94,UFF,Ghemical}]
[-mm {DistanceGeometry,SteepestDescent,ConjugateGradients}] [-co VDW_COMP]
[-i INTERACTING] [-ph PH] [-sa] [-a] [-o OUTPUT] [-m]
filename
############
# ARPEGGIO #
############
A library for calculating interatomic contacts in proteins
Dependencies:
- Python >= 3.6
- Numpy
- BioPython >= v1.80
- Open Babel >= 3.0.0
- gemmi >= 0.5.8
positional arguments:
filename Path to the file to be analysed.
options:
-h, --help show this help message and exit
-s SELECTION [SELECTION ...], --selection SELECTION [SELECTION ...]
Select the "ligand" for interactions, using selection syntax: /<chain_id>/<res_num>[<ins_code>]/<atom_name> or RESNAME:<het_id>. Fields can be omitted.
-sf SELECTION_FILE, --selection-file SELECTION_FILE
Selections as above, but listed in a file.
-wh, --write-hydrogenated
Write an output file including the added hydrogen coordinates.
-mh, --minimise-hydrogens
Energy minimise OpenBabel added hydrogens.
-ms MINIMISATION_STEPS, --minimisation-steps MINIMISATION_STEPS
Number of hydrogen minimisation steps to perform.
-mf {MMFF94,UFF,Ghemical}, --minimisation-forcefield {MMFF94,UFF,Ghemical}
Choose the forcefield to minimise hydrogens with. Ghemical is not recommended.
-mm {DistanceGeometry,SteepestDescent,ConjugateGradients}, --minimisation-method {DistanceGeometry,SteepestDescent,ConjugateGradients}
Choose the method to minimise hydrogens with. ConjugateGradients is recommended.
-co VDW_COMP, --vdw-comp VDW_COMP
Compensation factor for VdW radii dependent interaction types.
-i INTERACTING, --interacting INTERACTING
Distance cutoff for grid points to be 'interacting' with the entity.
-ph PH pH for hydrogen addition.
-sa, --include-sequence-adjacent
For intra-polypeptide interactions, include non-bonding interactions between residues that are next to each other in sequence; this is not done by default.
-a, --use-ambiguities
Turn on abiguous definitions for ambiguous contacts.
-o OUTPUT, --output OUTPUT
Define output directory.
-m, --mute Silent mode without debug info.
import json
import pandas as pd
Arpeggio работает с файлами в формате cif. Используйте данный вам pdbid отсюда.
!wget http://www.rcsb.org/pdb/files/1HKC.cif.gz
!gzip -d 1HKC.cif.gz
!ls
--2024-12-25 20:36:25-- http://www.rcsb.org/pdb/files/1HKC.cif.gz Resolving www.rcsb.org (www.rcsb.org)... 128.6.159.248 Connecting to www.rcsb.org (www.rcsb.org)|128.6.159.248|:80... connected. HTTP request sent, awaiting response... 301 Moved Permanently Location: https://www.rcsb.org/pdb/files/1HKC.cif.gz [following] --2024-12-25 20:36:25-- https://www.rcsb.org/pdb/files/1HKC.cif.gz Connecting to www.rcsb.org (www.rcsb.org)|128.6.159.248|:443... connected. HTTP request sent, awaiting response... 301 Moved Permanently Location: https://files.rcsb.org/download/1HKC.cif.gz [following] --2024-12-25 20:36:26-- https://files.rcsb.org/download/1HKC.cif.gz Resolving files.rcsb.org (files.rcsb.org)... 128.6.159.245 Connecting to files.rcsb.org (files.rcsb.org)|128.6.159.245|:443... connected. HTTP request sent, awaiting response... 200 OK Length: 195094 (191K) [application/octet-stream] Saving to: ‘1HKC.cif.gz’ 1HKC.cif.gz 100%[===================>] 190.52K --.-KB/s in 0.06s 2024-12-25 20:36:26 (3.31 MB/s) - ‘1HKC.cif.gz’ saved [195094/195094] 1HKC.cif condacolab_install.log sample_data
!mkdir ./arp-results3
По умолчанию ищутся контакты внутри всей структуры. При обозначении одной цепи как лиганда (в данном случае цепи А, если в структуре всего одна цепь уберите флаг -s) у записей выдачи появляются метки interacting_entities, см выдачу ниже.
!pdbe-arpeggio 1HKC.cif -ph 7.0 -wh -o ./arp-results3 -s /A//
INFO//20:36:27.157//Program begin. INFO//20:36:27.157//Selection perceived: ['/A//'] DEBUG//20:36:27.493//Loaded PDB structure (BioPython) ============================== *** Open Babel Warning in PerceiveBondOrders Failed to kekulize aromatic bonds in OBMol::PerceiveBondOrders DEBUG//20:36:28.061//Loaded MMCIF structure (OpenBabel) DEBUG//20:36:28.135//Mapped OB to BioPython atoms and vice-versa. DEBUG//20:36:28.499//Added hydrogens. DEBUG//20:36:28.731//Wrote hydrogenated structure file. Hydrogenation was by Arpeggio using OpenBabel defaults. DEBUG//20:36:33.274//Determined atom explicit and implicit valences, bond orders, atomic numbers, formal charge and number of bound hydrogens. DEBUG//20:36:33.375//Initialised SIFts. DEBUG//20:36:33.389//Determined polypeptide residues, chain breaks, termini DEBUG//20:36:33.389//Percieved and stored rings. DEBUG//20:36:33.392//Perceived and stored amide groups. DEBUG//20:36:33.406//Added hydrogens to BioPython atoms. DEBUG//20:36:33.426//Added VdW radii. DEBUG//20:36:33.444//Added covalent radii. DEBUG//20:36:33.459//Completed NeighborSearch. DEBUG//20:36:33.459//Assigned rings to residues. DEBUG//20:36:33.491//Made selection. DEBUG//20:36:34.045//Expanded to binding site. DEBUG//20:36:34.055//Flagged selection rings. DEBUG//20:36:34.072//Completed new NeighbourSearch. INFO//20:57:42.233//Program End. Maximum memory usage was 180.7 MB.
!ls ./arp-results3
1HKC_hydrogenated.mmcif 1HKC.json
Обратите внимание на типы взаимодействующих объектов (внутри выборки, между выборкой и чем-то ещё и тд)
Выдача в виде json-файла, воспользуемся пакетом json для парсинга и переведём в более человеко-читаемый вид таблицы. Также отфильтруем контакты нужного нам типа между выборкой (цепью А в данном случае) и остальной структурой (INTER)
with open('./arp-results3/1HKC.json', 'r') as js_file:
contacts = json.load(js_file)
output_dict = [x for x in contacts if (any(y in ['proximal','vdw','vdw_clash','clash'] for y in x['contact']) and (x['interacting_entities'] == 'INTRA_SELECTION') ) ] #
df = pd.json_normalize(output_dict)
df
| contact | distance | interacting_entities | type | bgn.auth_asym_id | bgn.auth_atom_id | bgn.auth_seq_id | bgn.label_comp_id | bgn.label_comp_type | bgn.pdbx_PDB_ins_code | end.auth_asym_id | end.auth_atom_id | end.auth_seq_id | end.label_comp_id | end.label_comp_type | end.pdbx_PDB_ins_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | [proximal] | 4.41 | INTRA_SELECTION | atom-atom | A | NH2 | 196 | ARG | P | A | CB | 132 | GLU | P | ||
| 1 | [proximal] | 4.88 | INTRA_SELECTION | atom-atom | A | NH2 | 196 | ARG | P | A | CG | 132 | GLU | P | ||
| 2 | [proximal] | 4.52 | INTRA_SELECTION | atom-atom | A | CB | 132 | GLU | P | A | CZ | 196 | ARG | P | ||
| 3 | [proximal] | 4.22 | INTRA_SELECTION | atom-atom | A | NH1 | 196 | ARG | P | A | CB | 132 | GLU | P | ||
| 4 | [proximal, weak_polar] | 3.48 | INTRA_SELECTION | atom-atom | A | CA | 132 | GLU | P | A | O | 128 | ASP | P | ||
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 37022 | [proximal] | 4.19 | INTRA_SELECTION | atom-atom | A | CE1 | 647 | PHE | P | A | CD | 644 | ARG | P | ||
| 37023 | [proximal] | 4.73 | INTRA_SELECTION | atom-atom | A | N | 646 | GLU | P | A | CG | 644 | ARG | P | ||
| 37024 | [proximal] | 4.93 | INTRA_SELECTION | atom-atom | A | NE | 644 | ARG | P | A | CD1 | 647 | PHE | P | ||
| 37025 | [proximal] | 4.21 | INTRA_SELECTION | atom-atom | A | CE1 | 647 | PHE | P | A | NE | 644 | ARG | P | ||
| 37026 | [proximal] | 4.83 | INTRA_SELECTION | atom-atom | A | O | 641 | ILE | P | A | CG | 644 | ARG | P |
37027 rows × 16 columns
Отфильтруем контакты только белок-белок, а также оставим только по 1 контакту между парой остатков.
df_protein_only = df.loc[(df['bgn.label_comp_type'] == 'P') & (df['end.label_comp_type'] == 'P')]
df_droped = df_protein_only.drop_duplicates(subset=['bgn.auth_seq_id', 'end.auth_seq_id'], keep='first')
df_droped.sort_values(by=['distance'])
| contact | distance | interacting_entities | type | bgn.auth_asym_id | bgn.auth_atom_id | bgn.auth_seq_id | bgn.label_comp_id | bgn.label_comp_type | bgn.pdbx_PDB_ins_code | end.auth_asym_id | end.auth_atom_id | end.auth_seq_id | end.label_comp_id | end.label_comp_type | end.pdbx_PDB_ins_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 23327 | [vdw_clash, polar] | 2.32 | INTRA_SELECTION | atom-atom | A | O | 448 | GLY | P | A | OG1 | 216 | THR | P | ||
| 26924 | [vdw_clash, polar] | 2.39 | INTRA_SELECTION | atom-atom | A | O | 256 | CYS | P | A | OG1 | 66 | THR | P | ||
| 31575 | [vdw_clash, polar] | 2.50 | INTRA_SELECTION | atom-atom | A | N | 572 | GLU | P | A | O | 570 | THR | P | ||
| 33100 | [vdw_clash, polar] | 2.50 | INTRA_SELECTION | atom-atom | A | O | 563 | PRO | P | A | N | 566 | ILE | P | ||
| 26989 | [vdw_clash, polar] | 2.53 | INTRA_SELECTION | atom-atom | A | OG1 | 259 | THR | P | A | O | 236 | ALA | P | ||
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 15677 | [proximal] | 5.00 | INTRA_SELECTION | atom-atom | A | CG | 639 | ASP | P | A | C | 636 | LEU | P | ||
| 11127 | [proximal] | 5.00 | INTRA_SELECTION | atom-atom | A | CB | 657 | ASP | P | A | C | 896 | GLY | P | ||
| 29790 | [proximal] | 5.00 | INTRA_SELECTION | atom-atom | A | N | 272 | ASP | P | A | NH2 | 279 | ARG | P | ||
| 13189 | [proximal] | 5.00 | INTRA_SELECTION | atom-atom | A | C | 500 | ARG | P | A | SD | 695 | MET | P | ||
| 11597 | [proximal] | 5.00 | INTRA_SELECTION | atom-atom | A | CB | 553 | VAL | P | A | CG2 | 545 | ILE | P |
7108 rows × 16 columns
Напишем функцию расчета split_value по номеру остатка. $SplitValue=\frac{intA}{extAB}\cdot\frac{intB}{extAB}$, где $intA$ – число пар контактирующих остатков из A, $intB$ – число пар контактирующих остатков из B, $extAB$ – число пар контактирующих остатков, один из A, а другой – из B.
def split_value(split_pos, df):
intA = len(df.loc[(df["bgn.auth_seq_id"] < split_pos) & (df["end.auth_seq_id"] < split_pos)])
intB = len(df.loc[(df["bgn.auth_seq_id"] >= split_pos) & (df["end.auth_seq_id"] >= split_pos)])
extAB = len(df) - intA - intB
if extAB != 0:
splitval = intA/extAB * intB/extAB
else:
splitval = 0
return splitval
Вычислим split_value для каждого остатка в вашем белке. Построим график split_value в зависимости от номера остатка.
first_res = df_droped[["bgn.auth_seq_id", "end.auth_seq_id"]].min().min()
last_res = df_droped[["bgn.auth_seq_id", "end.auth_seq_id"]].max().max()
split_data = [split_value(split_pos, df_droped) for split_pos in range(first_res, last_res)]
from matplotlib import pyplot as plt
plt.plot(range(first_res, last_res), split_data)
[<matplotlib.lines.Line2D at 0x7f1523318070>]

Определим номер остатка, соответствующий максимальному значению split_value. Остатки до него отнесём к домену A, остатки после него отнесём к домену B.
import numpy as np
[split_pos for split_pos in range(first_res, last_res)][np.argmax(split_data)]
465
Сравнение с доменами из SCOP и CATH¶
С помощью алгоритма DOMAK удалось определить два домена, в то время как и в SCOP, и в CATH для этой структуры выделено 4 домена. Однако разбиение на домены не одинаково. В SCOP каждый домен представлен подряд идущими остатками, в то время как в пространстве некоторые из них удалены на столько, что между ними распологается другой домен. В то время как в CATH разбиение на домены больше соответсвует пространственному расположению остатков, также часть α-спирали в ценре белка не отнесена ни к какому домену. Однако разбиение на домены по этой спирали справедливо как для предсказания, так и для обоих баз данных. При этом разбиение каждой половины белка на ещё два домена уже не столь тривиально, т.к. они сильно сближены, поэтому не было предсказано и различается в двух базах данных (рис. 1).

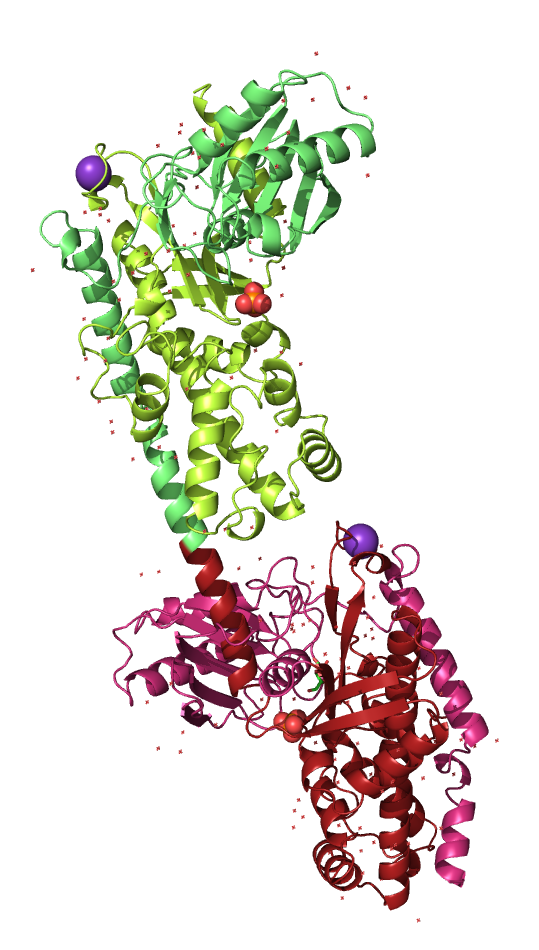

Поиск доменов в базе Interpro¶
Согласно данным из базы InterPro белок состоит из двух эволюционных доменов, причём разбиение так же происходит по центральной α-спирали (часть не отнесена ни к какому домену), что соотносится с предсказанием DOMAK.
| Границы доменов | ||||
|---|---|---|---|---|
| DOMAK | 16-465 | 465-914 | ||
| SCOP | 16-222 | 223-465 | 466-670 | 671-914 |
| CATH | 54-222;446-463 | 16-53;223-445 | 513-670;894-914 | 481-512;671-892 |
| InterPro | 16-458 | 464-906 | ||

Работа с разметкой вторичной структуры в ручном режиме¶
Я честно пытался разобраться как выполнить эту часть практикума, но мне не хватило компетенций ((
Главные выводы:
- сайт недоступен с территории России (при этом даже с VPN половина страниц всё ещё не открывается)
- поиск по PDBID, или UniProtID, или последовательности не даёт результатов
- поливина кнопок сайта открывают окно входа/регистрации, но при этом регистрация не работает