Сборка de novo
Получение чтений
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/008/SRR4240358/SRR4240358.fastq.gz
AC: SRR4240358
Организм: Buchnera aphidicola str. Tuc7 (Acyrthosiphon pisum)
Прибор: Illumina Genome Analyzer II
Тип: одноконцевые (SE)
Подготовка чтений программой trimmomatic
Удаление адаптеров
Я создала поддиректорию pr15, в которой находятся все данные для этого практикума. Скопировала в неё файлы из /mnt/scratch/NGS/adapters и объединила их с помощью команды
cat * > adapters.faДалее вырезала адаптеры:
TrimmomaticSE -threads 10 SRR4240358.fastq.gz SRR4240358_without_adapters.fastq.gz ILLUMINACLIP:adapters.fa:2:7:7 2> log.txt
В файл log.txt была записана информация о работе программы. Из него узнала, что 1,66% последовательностей оказались адаптерами и были удалены.
Удаление нуклеотидов с низким качеством
TrimmomaticSE -threads 10 -phred33 SRR4240358_without_adapters.fastq.gz trimm_SRR4240358.fastq.gz TRAILING:20 MINLEN:32 2> trimm_log.txt
TRAILING:20 устанавливает, что будут удаляться нуклеотиды с качеством меньше 20. MINLEN:32 удаляет чтения длиной меньше 32 нуклеотидов.
В файле с логами можно увидеть, что было удалено 2352447 (22.69%) чтений.
Размеры файлов:
Изначальный файл с чениями – 493М
Без адаптеров – 485М
После триммирования – 357М
Получение k-меров
velveth velveth_out/ 31 -fastq.gz -short trimm_SRR4240358.fastq.gz
velveth_out/ – директория, куда будут помещены файлы с результатом работы программы;
31 – длина k-меров;
-fastq.gz – опция, задающая формат файла с чтениями;
-short – короткие непарные чтения
trimm_SRR4240358.fastq.gz – файл с чтениями, подающийся на вход программе
Сборка k-меров
velvetg velveth_out/
velveth_out/ – папка с файлами, полученными в результате работы velveth.
Из вывода в stdout прочитала, что N50 = 8600; максимальная длина контига – 19821.
В файле stats.txt находятся данные о контигах в виде таблицы с разделением табуляцией. Длина находится во втором столбце. Длины трёх самых длинных контигов нашла с помощью команды
cut -f2 stats.txt | sort -n | tail -n 3
Далее открыла stats.txt и с помощью поиска по файлу нашла ID и покрытия для этих трёх самых длинных контигов:
| ID | Длина | Покрытие |
|---|---|---|
| 56 | 19821 | 29.475859 |
| 34 | 18714 | 29.922678 |
| 40 | 16436 | 30.793624 |
Чтобы найти контиги с аномальными покрытиями, отсортировала стобец с покрытиями с помощью команды
cut -f6 stats.txt | sort -n > cov.txt
Есть контиги с аномально большими и аномально маленькими покрытмиями. Вот некоторые из них:
| ID | Покрытие | Длина |
|---|---|---|
| 362 | 1.333333 | 6 |
| 359 | 1.142857 | 7 |
| 342 | 1.750000 | 8 |
| ID | Покрытие | Длина |
|---|---|---|
| 136 | 111576.000000 | 1 |
| 142 | 552.636364 | 11 |
| 140 | 458.000000 | 1 |
Видно, что длины всех контигов с аномальными покрытиями меньше 31 (длина k-меров). Такие контиги не попадают в contig.fa. Контиг, у которого покрытие выбивается больше всего (111576.000000) имеет длину 1 нуклеотид, поэтому неудивительно, что у него такое большое покрытие
Анализ
Я разбила файл на отдельные контиги с помощью seqretsplit и затем осуществила megablast с контигами с наибольшей длиной и хромосомой Buchnera aphidicola.
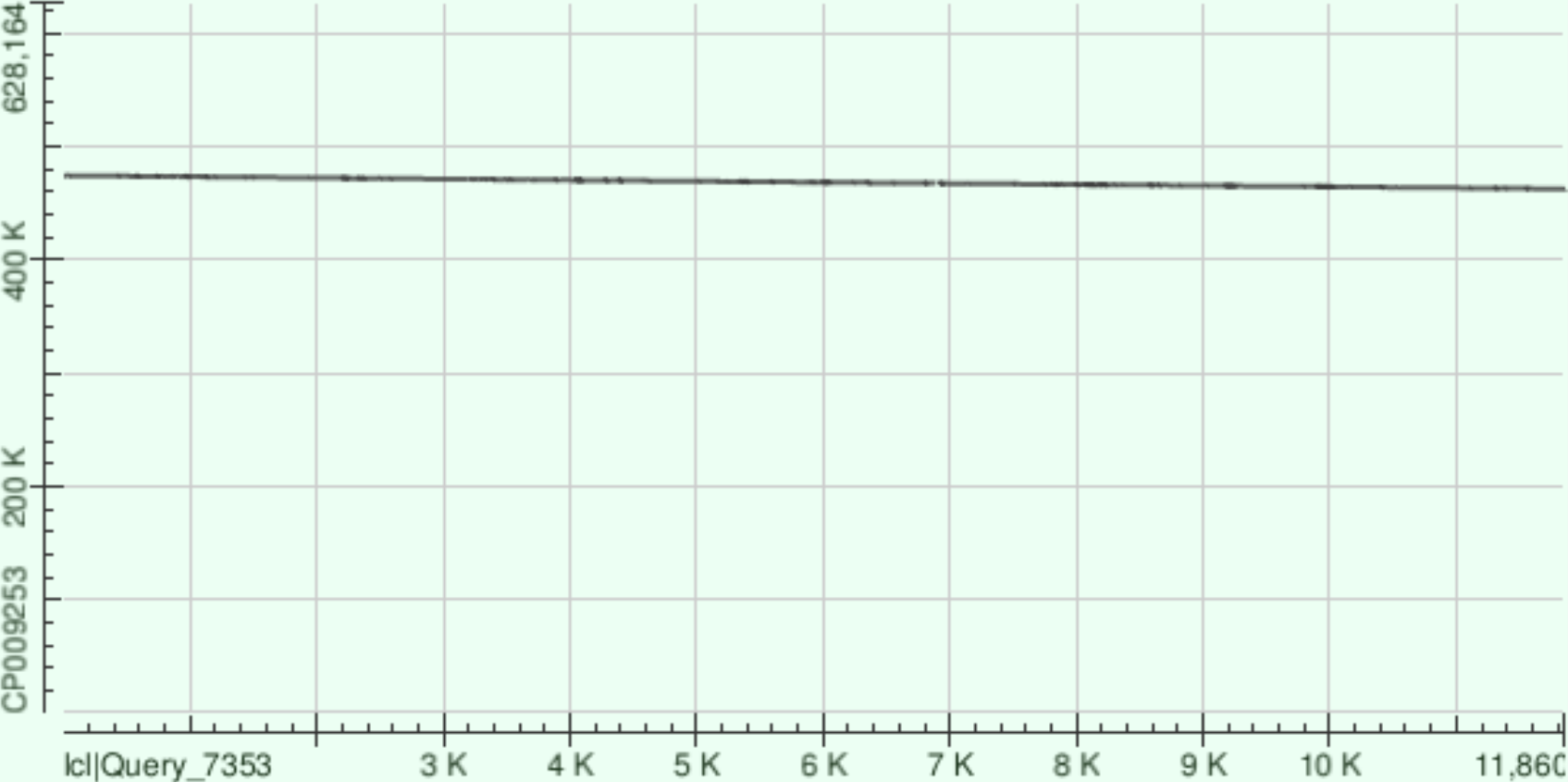
Самый длинный контиг
Было найдено три гомологичных участка:
| Координаты хромосомы | Гэпы | Процент идентичности |
|---|---|---|
| 496111-500325 | 154 | 75% |
| 500370-508806 | 351 | 76% |
| 510438-514772 | 77 | 81% |
Видно, что между вторым и третьим участком находится большой негомологичный участок
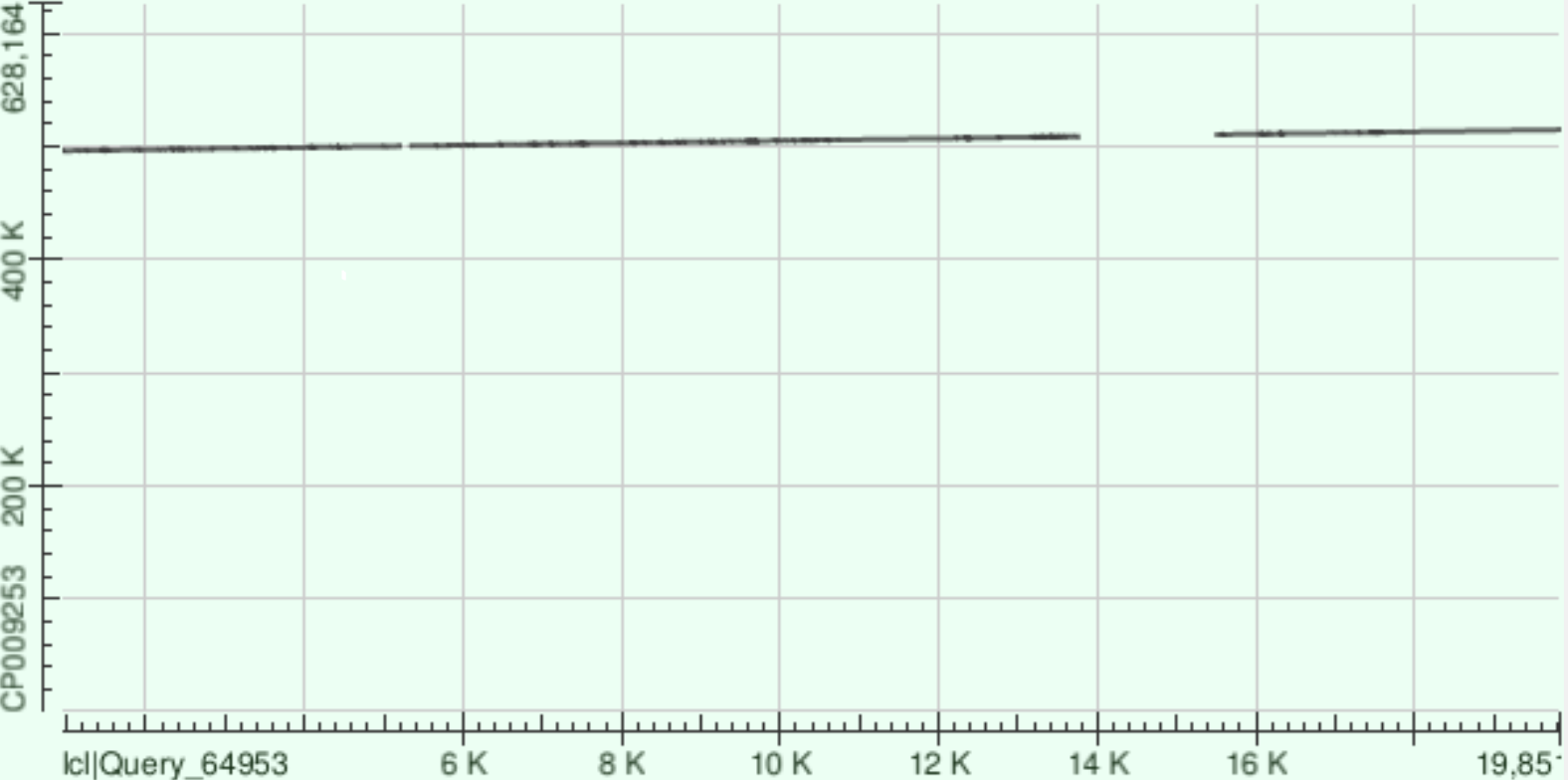
Второй по длине контиг
Было найдено 6 гомологичных участков:
| Координаты хромосомы | Гэпы | Процент идентичности |
|---|---|---|
| 8599-11103 | 50 | 78% |
| 13994-14465 | 9 | 82% |
| 14727-17919 | 86 | 76% | 17962-20171 | 30 | 85% |
| 20358-22183 | 51 | 82% | 23067-26764 | 140 | 78% |
На DotPlot видно, что между гомологичными участками происходили делеции. Между первым и вторым находится большой негомологичный участок.
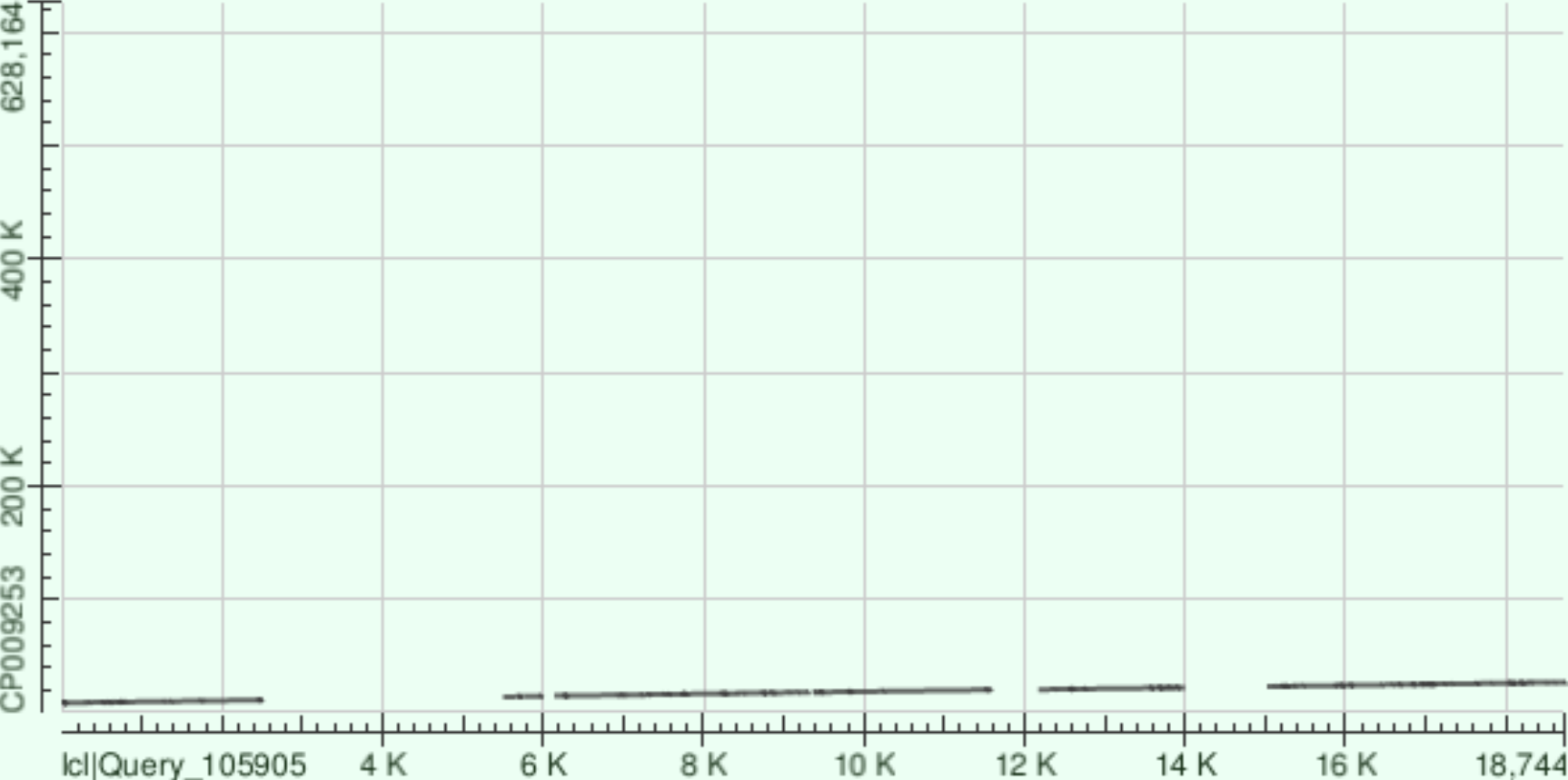
Третий по длине контиг
Было найдено 2 гомологичных участка:
| Координаты хромосомы | Гэпы | Процент идентичности |
|---|---|---|
| 467421-462496 | 162 | 77% |
| 474242-467412 | 204 | 77% |
На DotPlot наклон отрицательный, значит контиг был перевёрнут