AlphaFold. DOMAK.
Задание 1B (Helices) с последовательностями B9
В первом задании я выбрала вариант B (Helices). В этом задании дано три последовательности. SeqA, seqB и seqC практически идентичны за исключением трёх аминокислот в seqB, которые заменены на пролины, а в seqC на пролины заменены 12 аминокислот, включая те же три. Атом азота пролина входит в состав жёсткого кольца, что исключает возможность вращения вокруг связи N-CH. Кроме того, атом азота пролина, образующего пептидную связь с другой аминокислотой, не протонирован. В результате пролин не способен образовывать водородную связь и структура альфа-спирали нарушается. Обычно в этом месте возникает петля или изгиб.
Я выбрала последовательности B9. Их длина - 262 а.о. В seqB остатки 141-143 заменены на Pro, в seqC – остатки 137-148. С помощью поиска в RCSB я выяснила, что это мономер белка 3ONQ.
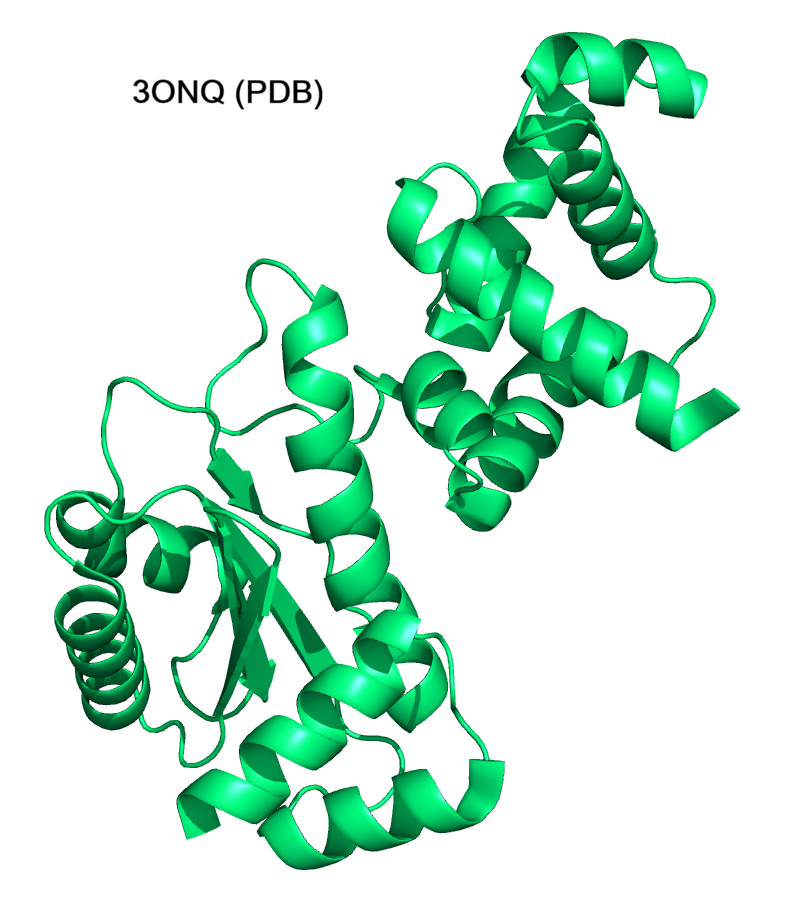

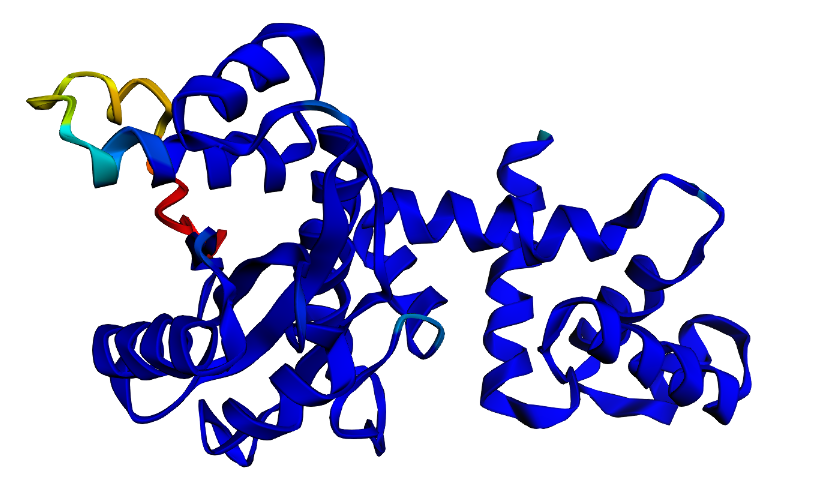
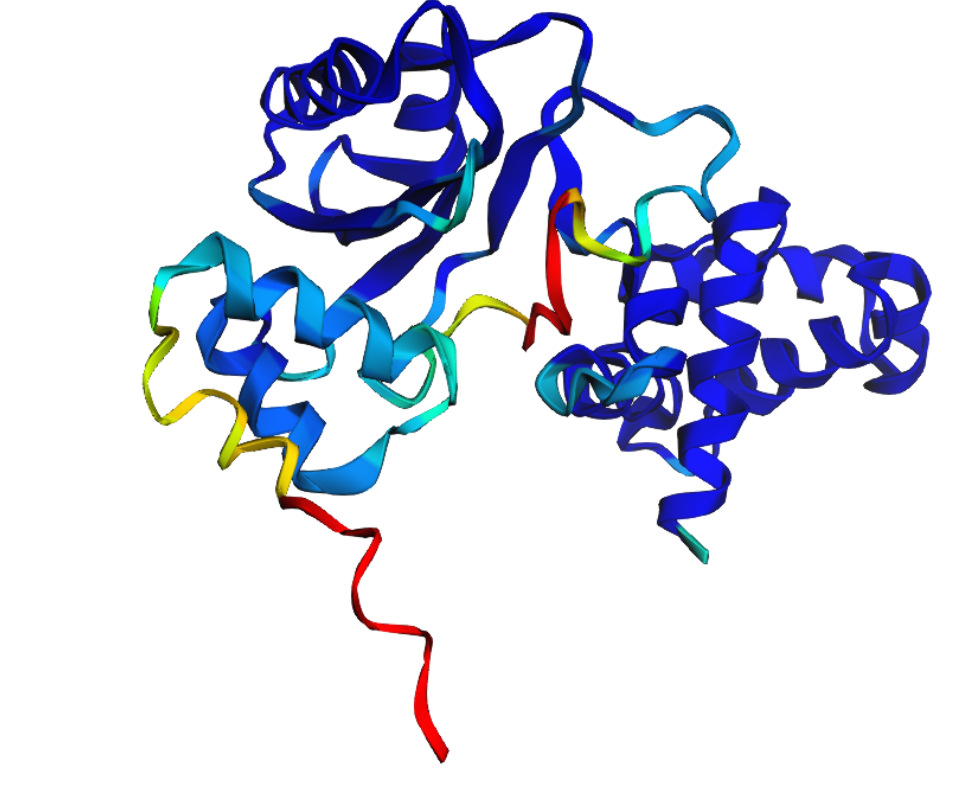
В PDB представлена структура гомотетрадимера – "Регулятор экспрессии поликетидсинтазы BAD_0249, выделенный из Bifidobacterium adolescentis". На Рис 1 видно, что предсказанная структура хорошо выравнивается со структурой из PDB, а на Рис 3 показано, что у seqA почти по всей структуре pIDDT > 0,9.
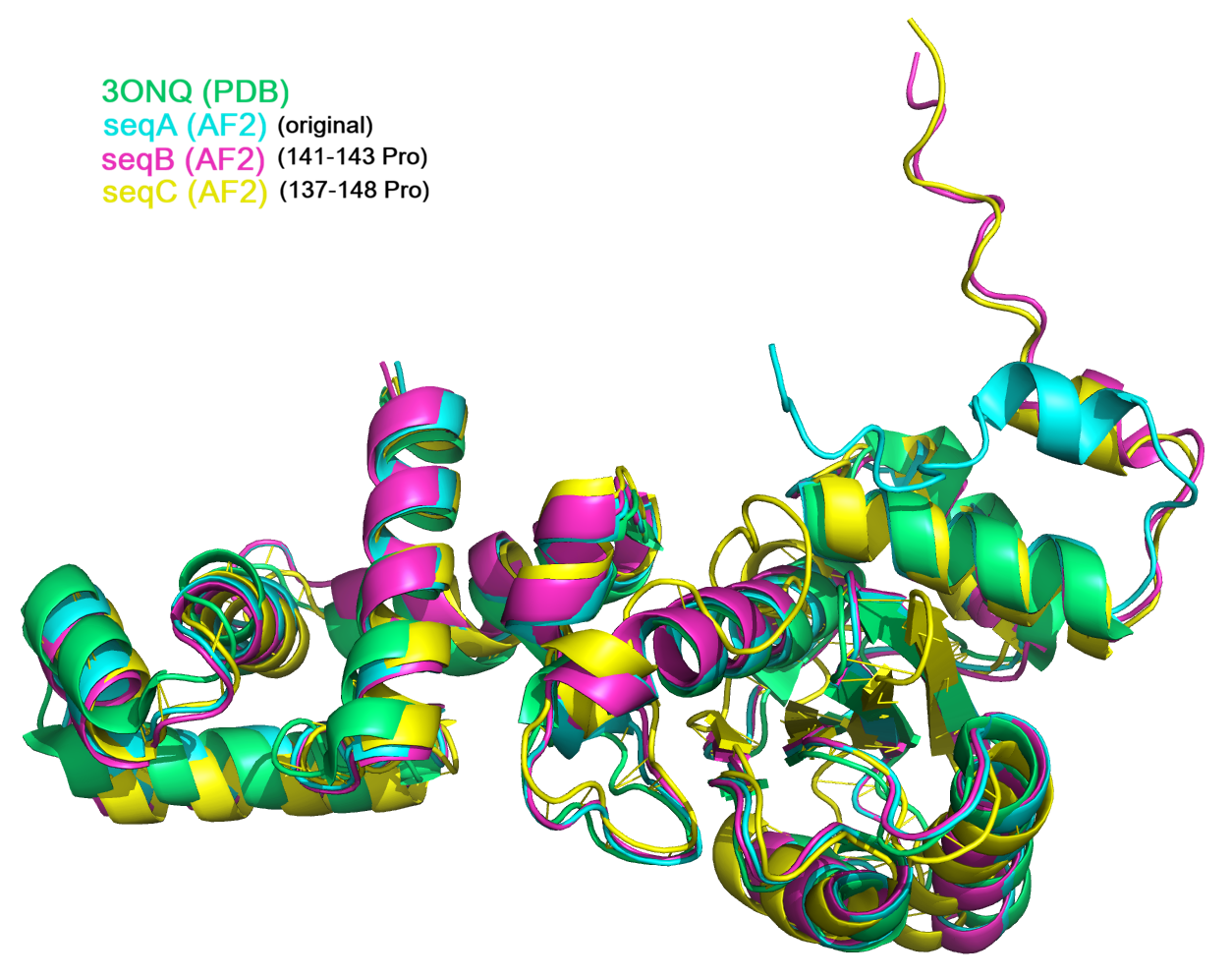
На Рис 2 представлено выравнивание структуры из PDB со всеми тремя предсказанными структурами.Все они хорошо выравниваются за исключением участка где у seqA (жёлтый) петля, а у всех остальных структур – альфа-спираль. Именно в этом месте находится участок с 12 пролинами. У всех предсказаний по всей области pIDDT > 0,9 (high confidence) кроме N-конца и того самого участка с петлёй у seqC.

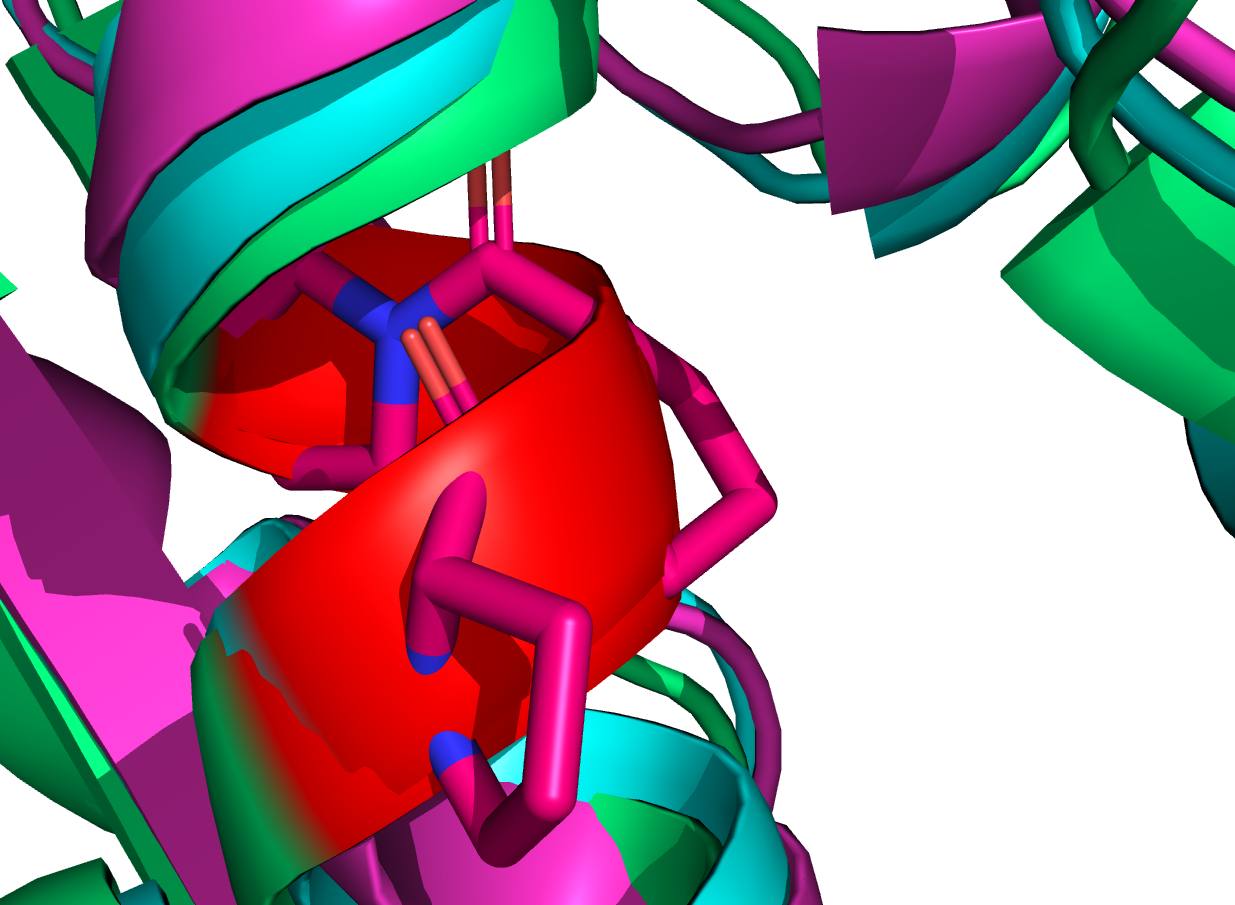
На Рис 6 показано влияние замены трёх аминокислот 141-143 на пролины. В этом случае альфа-спираль не нарушается. Видимо, в контексте данной последовательности трёх пролинов недостаточно для нарушения альфа-спирали.
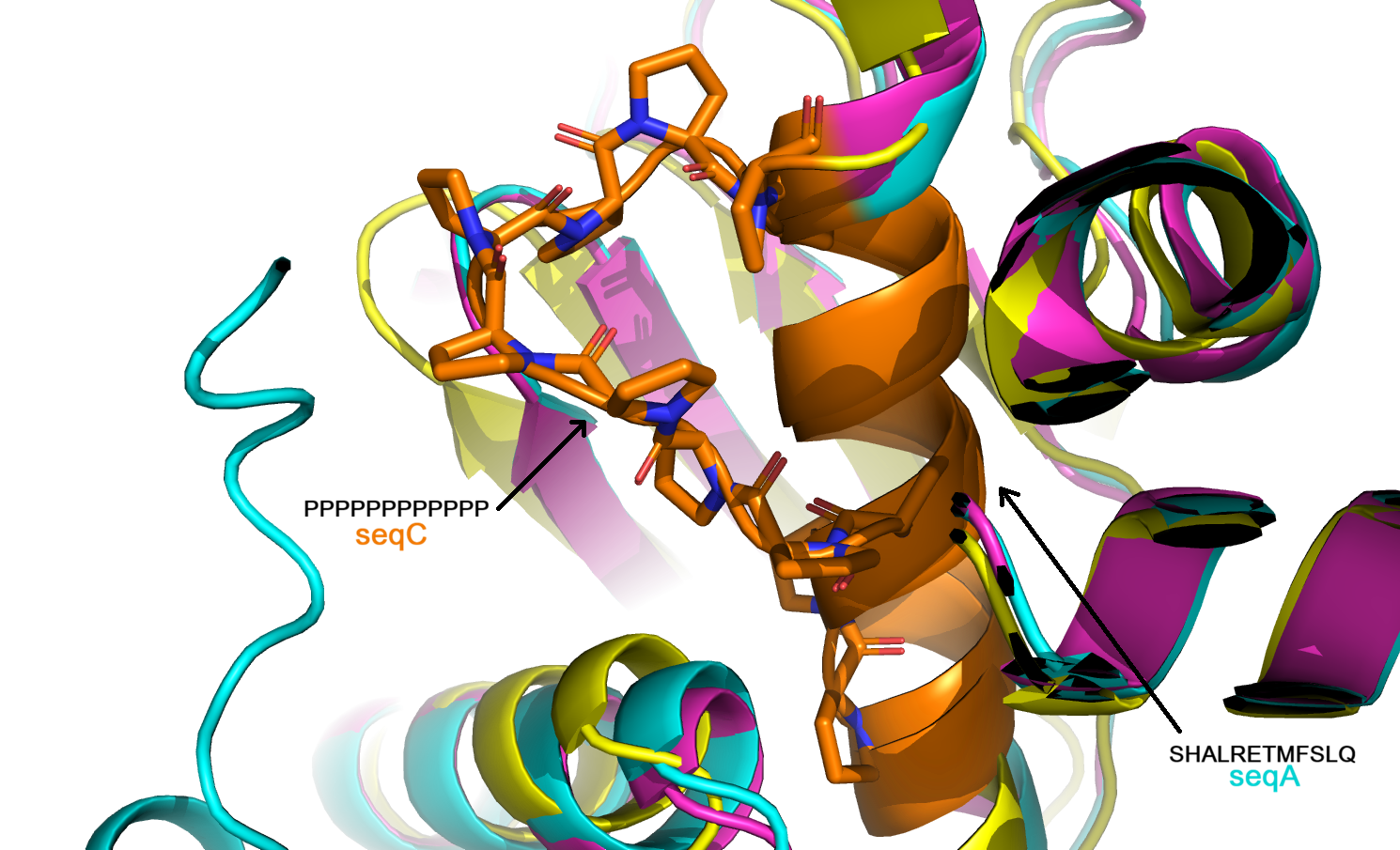
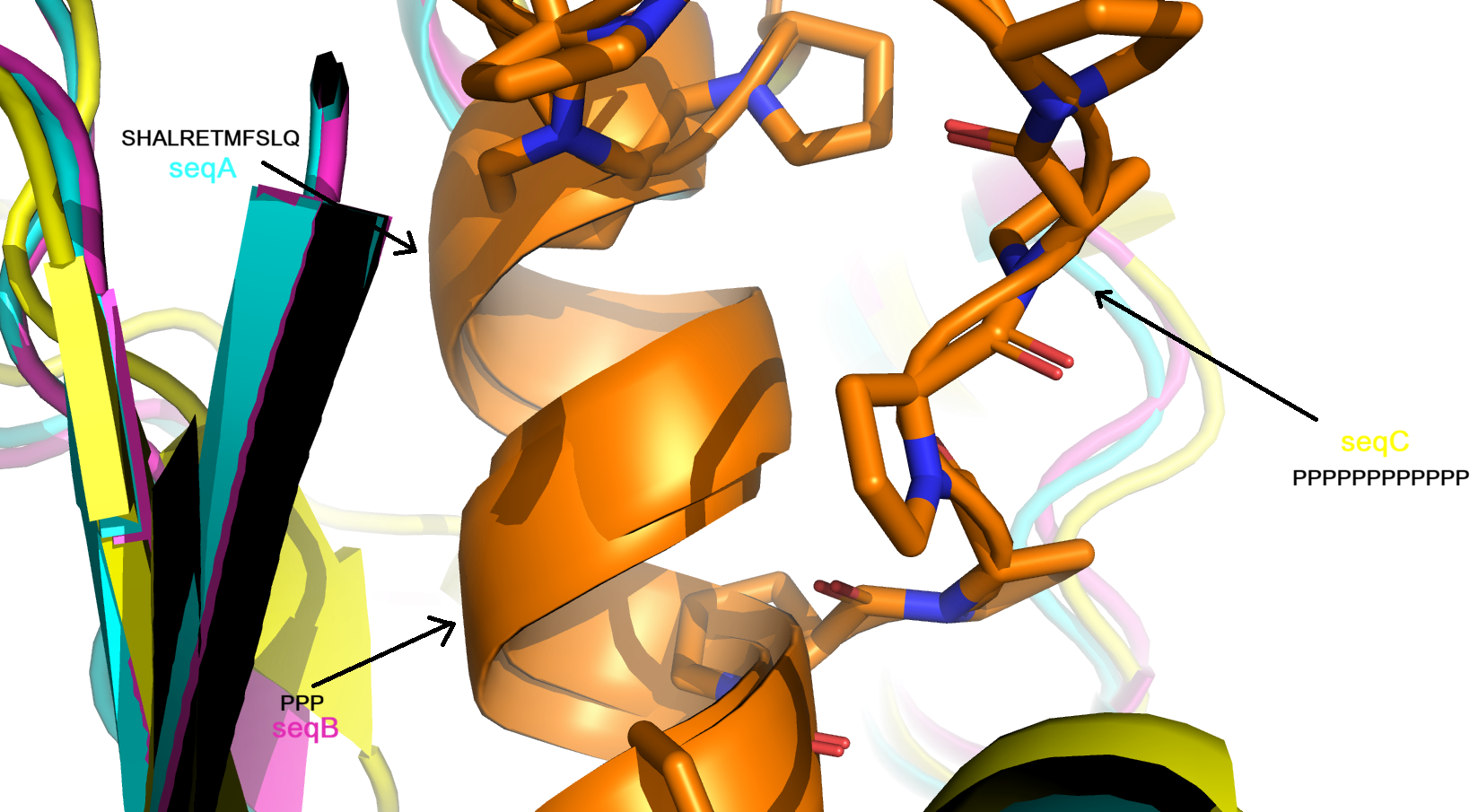
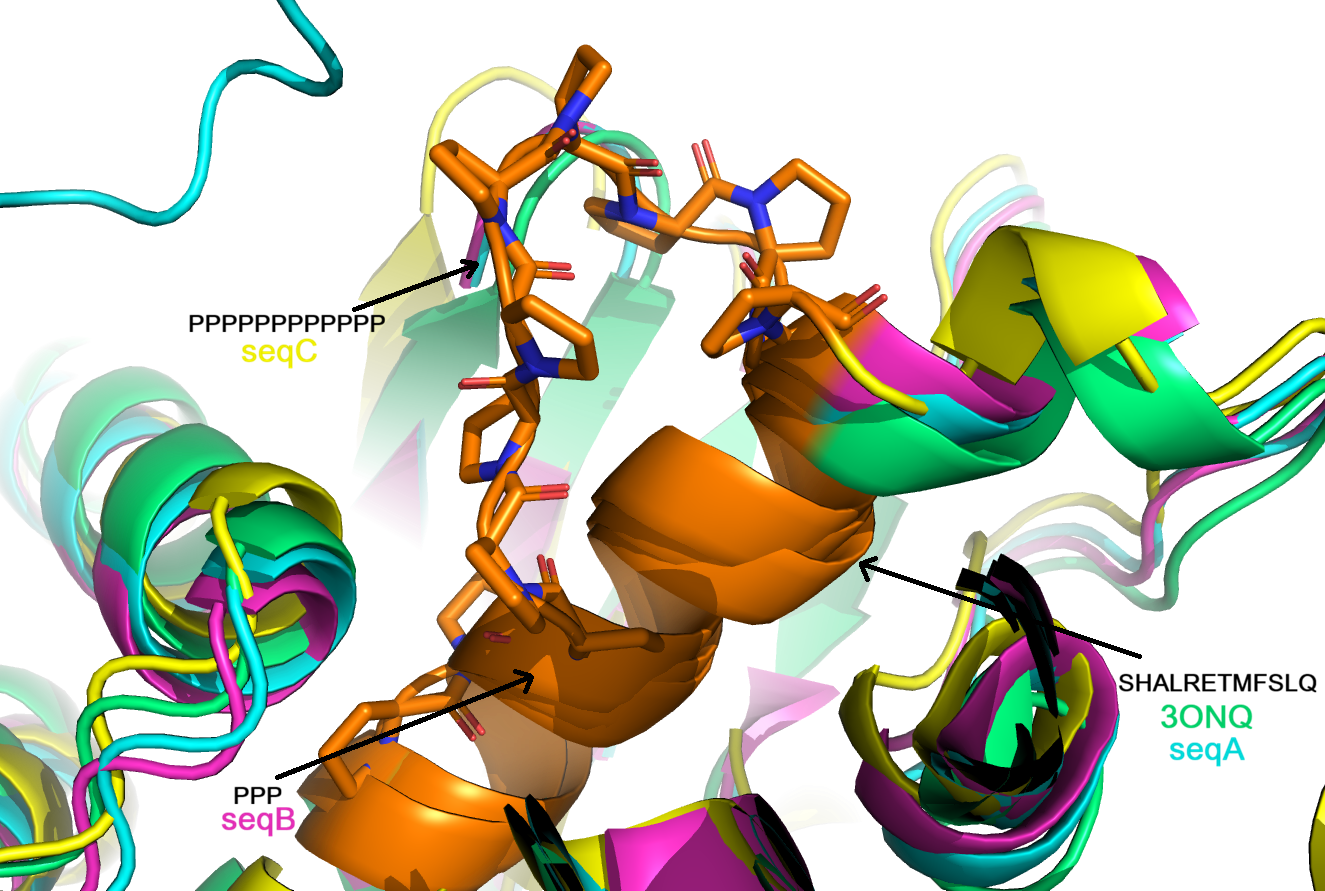
На Рис 7-8 представлено выравнивание структуры из PDB и трёх предсказанных структур, на котором выделены остатки 137-148, заменённые на Pro в seqC. Видно, что в структуре PDB и предсказаниях для seqA и seqB на этом месте находится альфа-спираль. У seqC на этом участке петля. Это ожидаемо, так как такой длинный участок из пролинов не может образовывать альфа-спираль по причинам, описанным выше.
Вывод
В целом, AlphaFold хорошо предсказал структуру, она соответствует структуре из PDB. В месте замены 12 аминокислот на пролины ожидаемо альфа спираль не сворачивается, так как пролины не могут образовывать водородные связи, на этом месте находится петля. Однако замена на три пролина почему-то не нарушает структуру. Видимо, AF2 считает, что в данном окружении и в контексте данной последовательности образование альфа-спирали возможно и более энергетически выгодно.
Задание 2-3. DOMAK
Ссылка на Colab. В этом задании я работала с белком 2C78, цепь A. Я применила алгоритм для разбиения белка на два домена. В результате получился следующий график:
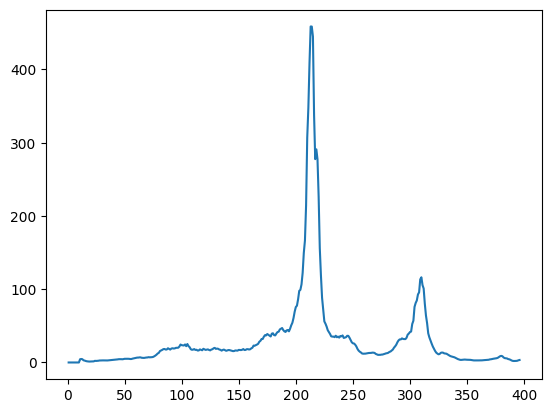
20 позиций с лучшим split_value: 212, 213, 214, 211, 210, 215, 209, 217, 216, 218, 219, 208, 207, 220, 206, 309, 308, 307, 310, 221
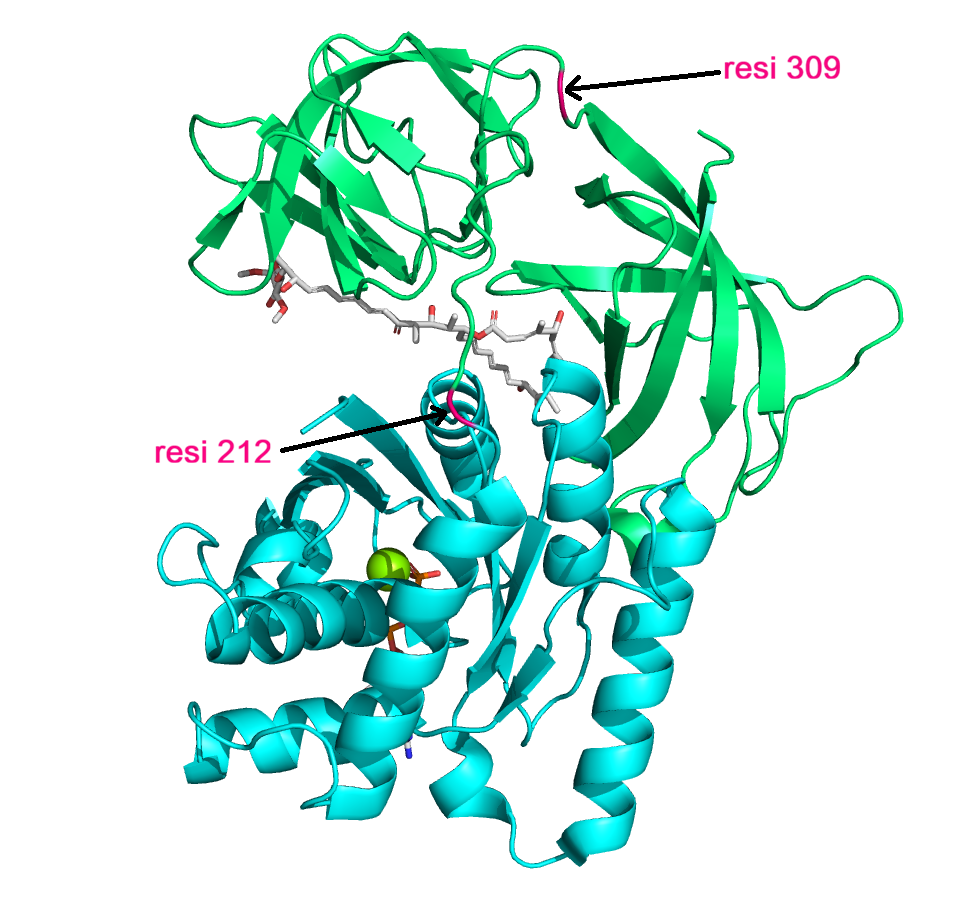
Это расположение певого пика 206-221 и частично второй пик, позиция с наилучшим split_value – 212. Посмотрим, как это выглядит на структуре.
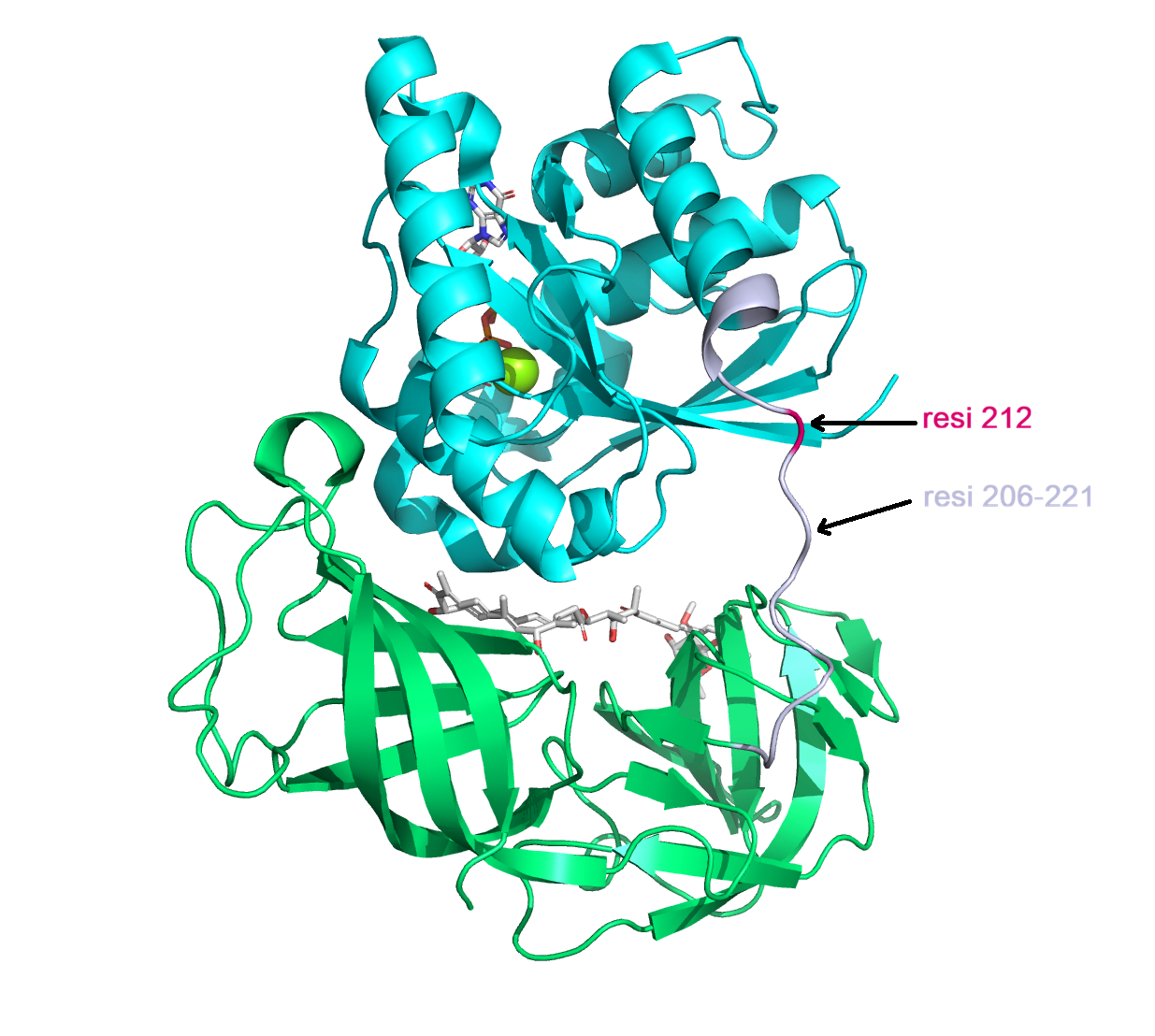
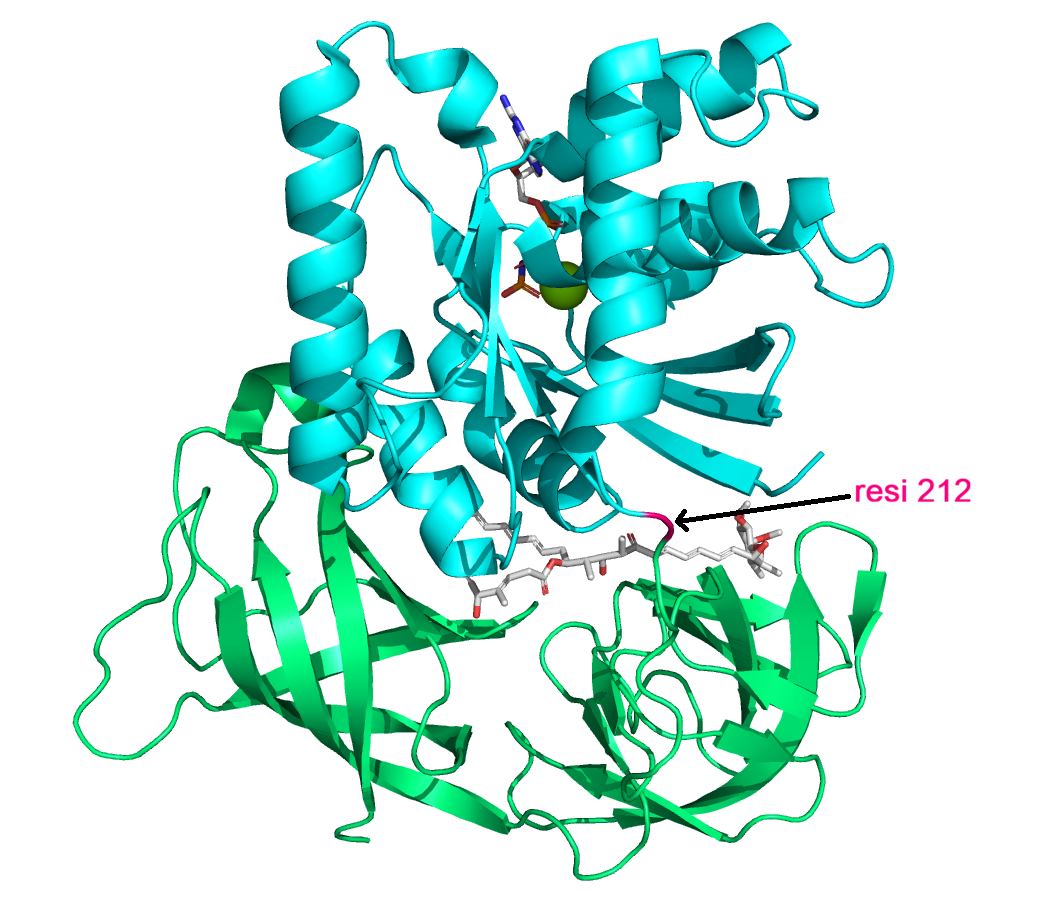
Посмотрим также на второй пик – 309. В целом, оба варианта выглядят правдоподобно, находятся вне вторичной структуры, хорошо визуально делят белок на структурные домены и находятся на достаточном расстоянии друг от друга. Возможно, здесь имеет место деление на три домена.
Теперь рассмотрим определённые для нашего белка структурные домены из баз SCOP и CATCH.
SCOP
SCOP также разделяет на три домена: 9-212 (P-loop containing nucleoside triphosphate hydrolases), 213-312 (EF-Tu/eEF-1alpha/eIF2-gamma C-terminal domain) и 313-405 (Translation proteins). Это очень похоже на предсказание DOMAK: первая позиция, разделяющая домены, полностью совпадает, а вторая отличается всего на 3 позиции.

CATCH
CATCH аналогично разбивает белок на три домена: 9-213 (P-loop containing nucleoside triphosphate hydrolases), 216-307 (Translation factors) и 311-404 (Translation factors). Также почти полностью совпадает с результатами DOMAK

Можно сделать вывод о том, что DOMAK хорошо предсказал разбиение на домены. Он правильно предсказал количество доменов, а также границы с погрешностью в несколько единиц, что очень хорошо.