Семи-эмперические вычисления: MOPAC.
Цель выполнения задания - постепенное освоение возможностей MOPAC, как пакета для оптимизации структуры молекул и расчёта некоторых свойств.
1. Оптимизация структуры порфирина
Сначала был создан файл 1.smi, содержащий аннотацию порфирина в виде SMILES (которая была получена на сайте ChEBI)и название молекулы.
На основе этого описания c помощью программы из openbabel была построена 3D-структура порфирина:
obgen 1.smi > 1.molС помощью программы PyMol были удалены ненужные атомы водорода (рисунок 1), полученная структура была сохранена в файле 1.pdb.
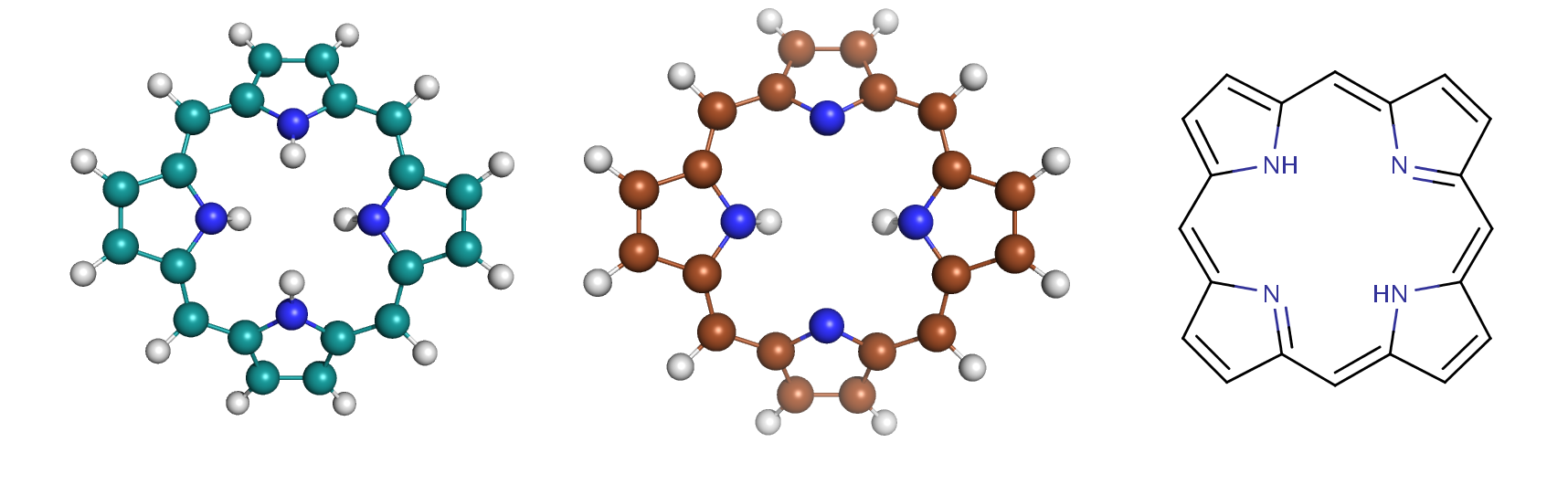
Рисунок 1. Структура порфирина, полученная с помощью программы из openbabel (слева), та же молекула после удаления ненужных атомов водорода (в центре), изображение структуры порфирина, полученное на сайте ChEBI.
babel -ipdb myfile.pdb -omop 1_opt.mop -xk "PM6"В данном случае мы провели оптимизацию с параметризацией pm6.
Далее запустили MOPAC, а затем переформатировали результат в .pdb-файл.
MOPAC2009.exe 1_opt.mop babel -imopout 1_opt.out -opdb 1_opt.pdbСравенение исходной и оптимизированной структур приведено на рисунке 2. Видно, что оптимизированная структура является плоской, в отличие от исходной.
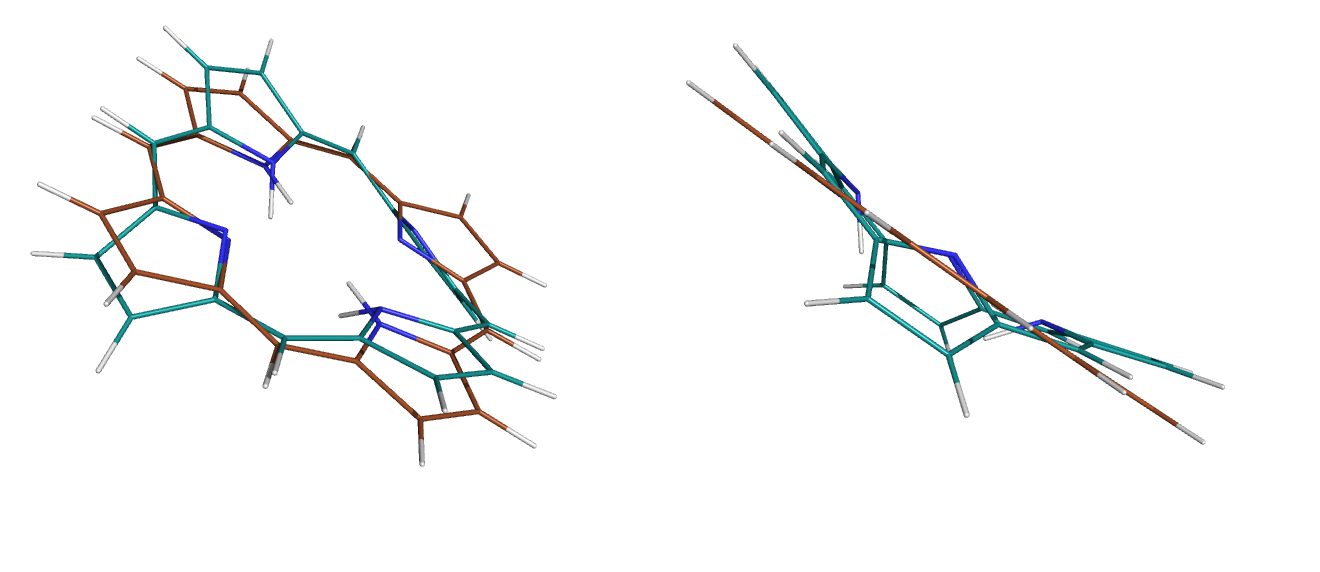
Рисунок 2. Исходная не плоская структура порфирина (изображена голубым цветом), и оптимизированная плоская структура (изображена коричневым цветом).
babel -ipdb myfile.pdb -omop 1_opt.mop -xk "AM1"На рисунке 3 изображены новая оптимизированная структура, и исходная структура порфирина.
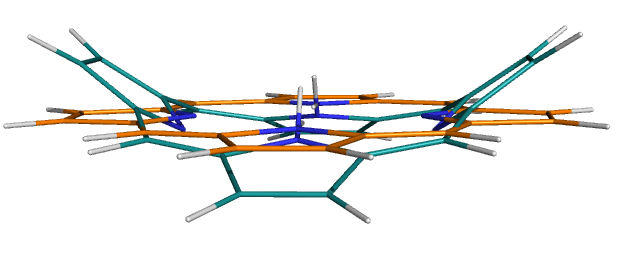
Рисунок 3. Исходная не плоская структура порфирина (изображена голубым цветом), и оптимизированная (с параметром AM1) плоская структура (изображена оранжевым цветом).
Рисунок 4. Сравнение структур, оптимизированных с параметрами PM6 (изображена коричневым цветом) и AM1 (изображена оранжевым цветом).
2. Расчет возбужденных состояний порфирина
Для того, чтобы оценить спектр поглощения порфирина, рассчитаем возбужденные состояния молекулы. Для расчёта возбуждённых состояний в копию .mop файла из предыдущего задания 1_spec.mop добавили строку в конец файла:cis c.i.=4 meci oldgeo For porphyrin spectrumДалее запустили MOPAC аналогично первому заданию. В конце выходного файла 1_spec.out можно найти значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.914512 1.914512 1 TRIPLET ????
3 2.267232 2.267232 2 SINGLET ????
4 2.465052 2.465052 2 TRIPLET ????
5 2.826208 2.826208 3 TRIPLET ????
6 3.365105 3.365105 4 TRIPLET ????
7 3.391955 3.391955 3 SINGLET ???? 0.2079 0.2334 0.0006
8 3.669444 3.669444 4 SINGLET ???? 2.3546 2.0779 0.0039
9 3.872312 3.872312 5 SINGLET ???? 1.5671 1.7688 0.0052
Из полученных данных об энергии можем посчитать длины волн: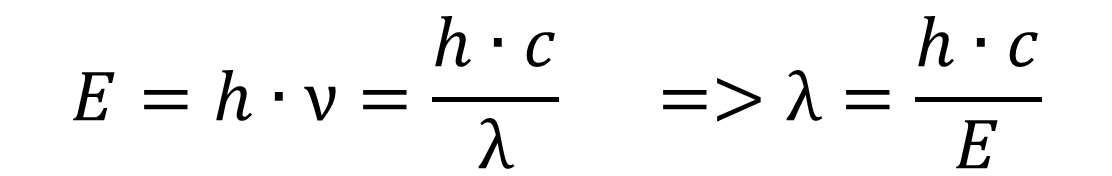 .
.Для энергии, измеренной в эВ, и длины волны, измеренной в нм:
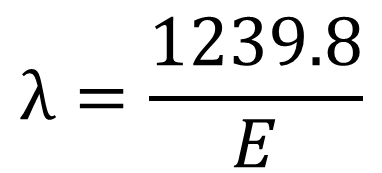 .
.Ниже приведены полученные значения длин волн, округленные до целого значения и отсортированные по возрастанию (длина волны указана в нм):
320; 337; 365; 368; 438; 503; 546; 647. На рисунке 5 приведено схематичное изображение спектра поглощения порфирина. Виден пик в УФ части спектра и несколько областей поглощения в видимой части спектра.

Рисунок 5. Схематичное изображение спектра поглощения молекулы порфирина.
3. Определение геометрии молекулы
Для молекулы O=C1C=CC(=O)C=C1 (хинон), с помощью шагов, аналогичных выполненным в задании 1, были получены структуры. Сравнение структур, полученных с помощью obgen и MOPAC, представлено на рисунке 6.
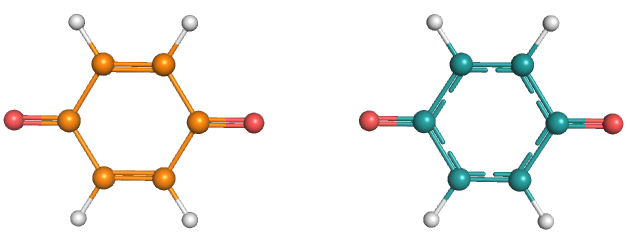
Рисунок 6. Структуры хинона, полученные с помощью obgen (слева, изображена оранжевым цветом) и MOPAC (справа, изображена голубым цветом).
Далее была определена геометрия дианиона молекулы хинона. Для этого сначала в файл .mop была вставлена первая строчка CHARGE=-2 и поставлены отрицательные заряды у атомов кислорода:
PM6 CHARGE=-2 quinone.pdb C 2.11000 1 -0.04100 1 -0.19000 1 ... O(-) 0.92800 1 -0.04700 1 -0.50300 1 O(-) 6.06800 1 -0.02300 1 0.85800 1 H 2.28400 1 2.13700 1 -0.14400 1 ...Далее уже привычным образом был получен файл quinone_dian_opt.pdb, содержащий структуру дианиона хинона. Сравнение молекул хинона и его дианиона представлено на рисунке 7.
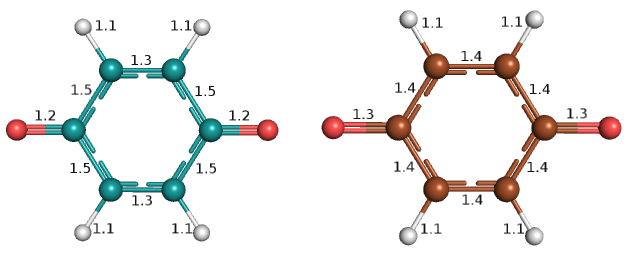
Рисунок 7. Структура хинона (слева, изображена голубым цветом) и его дианинона (справа, изображена коричневым цветом).
4. Тиминовые димеры
Ультрафиолет может превращать тимины в тиминовые димеры, а ДНК-фотолиаза при облучении ультрафиолетом востановливает основания тиминов до нормального состояния. Изначально нам дана структура тиминового димера (рисунок 8).
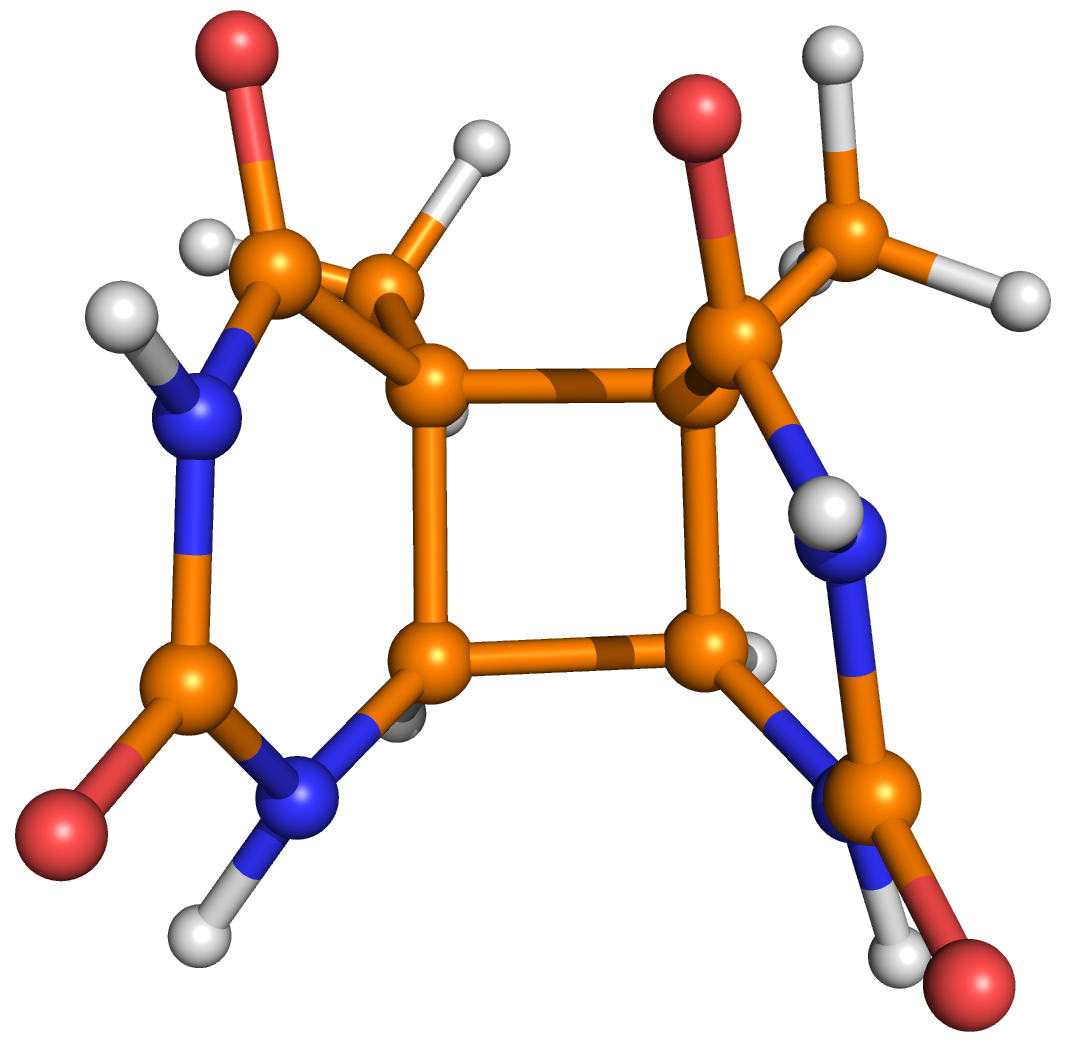
Рисунок 8. Структура тиминового димера.
Сначала проводим оптимизацию геометрии этого димера при заряде системы 0:
babel -ipdb td.pdb -omop td1.mop -xk "PM6" MOPAC2009.exe td1.mop babel -imopout td1.out -opdb td1.pdbНа рисунке 9 показана полученная оптимизированная структура.
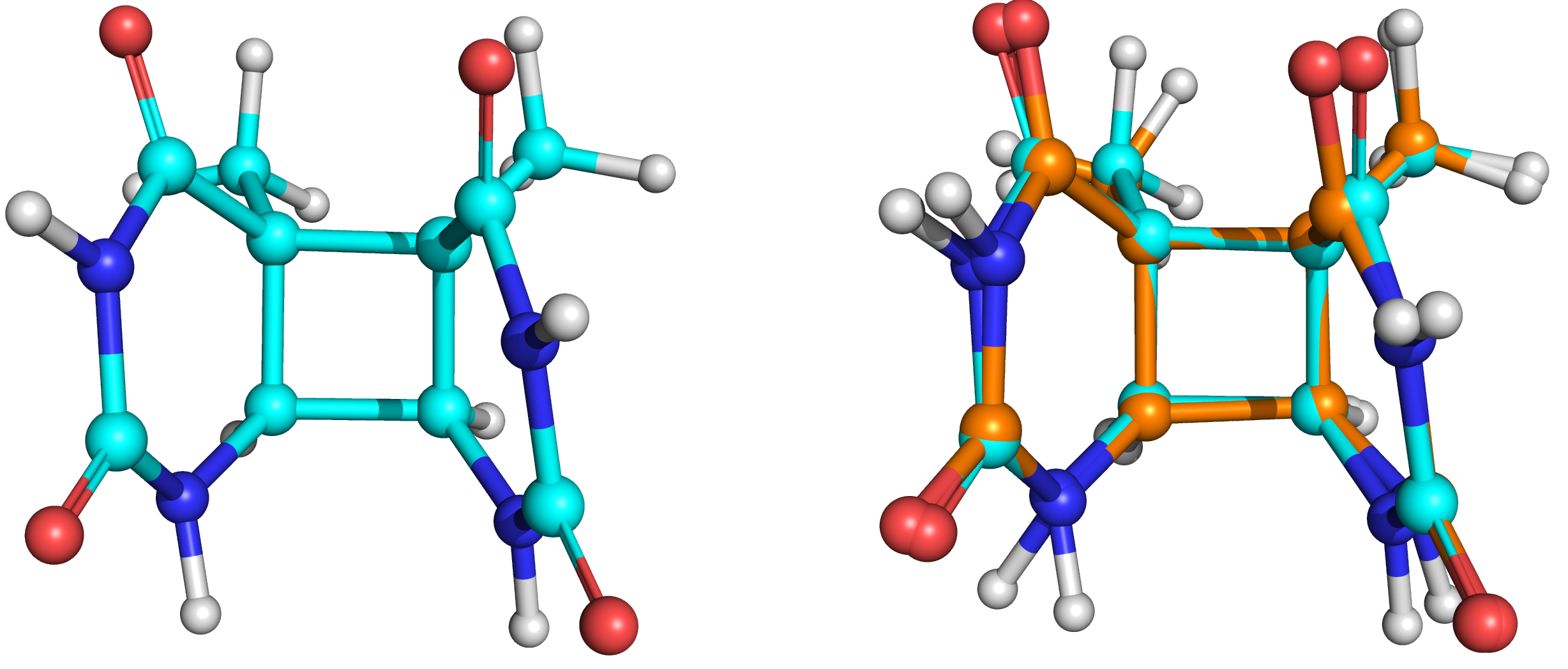
Рисунок 9. Оптимизированная структура димера при заряде системы 0. Справа показано наложение неоптимизированной структуры (оранжевый цвет) и оптимизированной (голубой цвет). Слева изображена оптимизированная структура.
babel -ipdb td1.pdb -omop td2.mop -xk "PM6" sed -i 's/PM6/PM6 CHARGE=+2/' td2.mop MOPAC2009.exe td2.mop babel -imopout td2.out -opdb td2.pdbПолученная структура показана на рисунке 10.
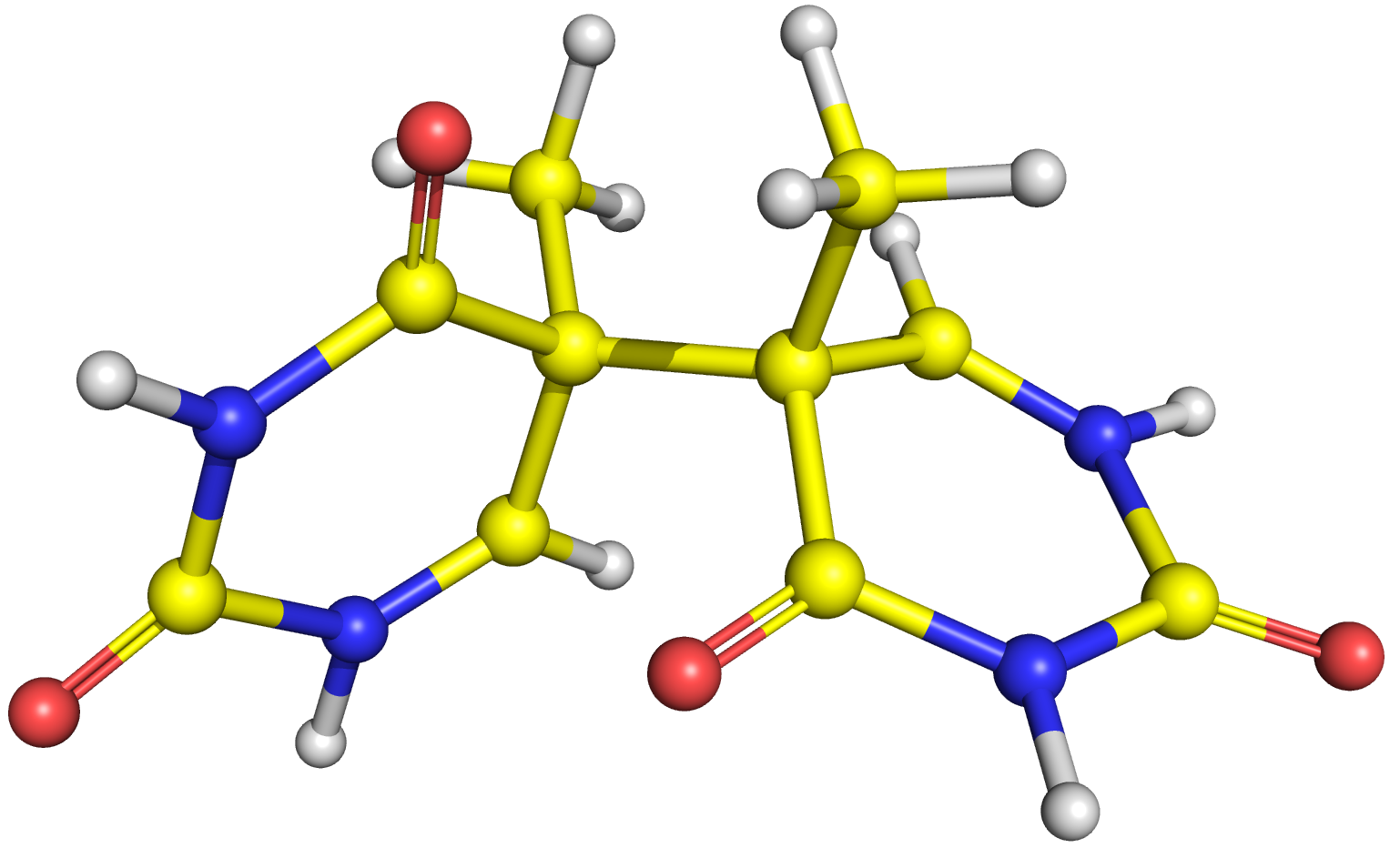
Рисунок 10. Оптимизированная структура димера при заряде системы +2. Перехода димера в тимины не наблюдается.
babel -ipdb td2.pdb -omop td3.mop -xk "PM6" MOPAC2009.exe td3.mop babel -imopout td3.out -opdb td3.pdbПолученная структура показана на рисунке 11.
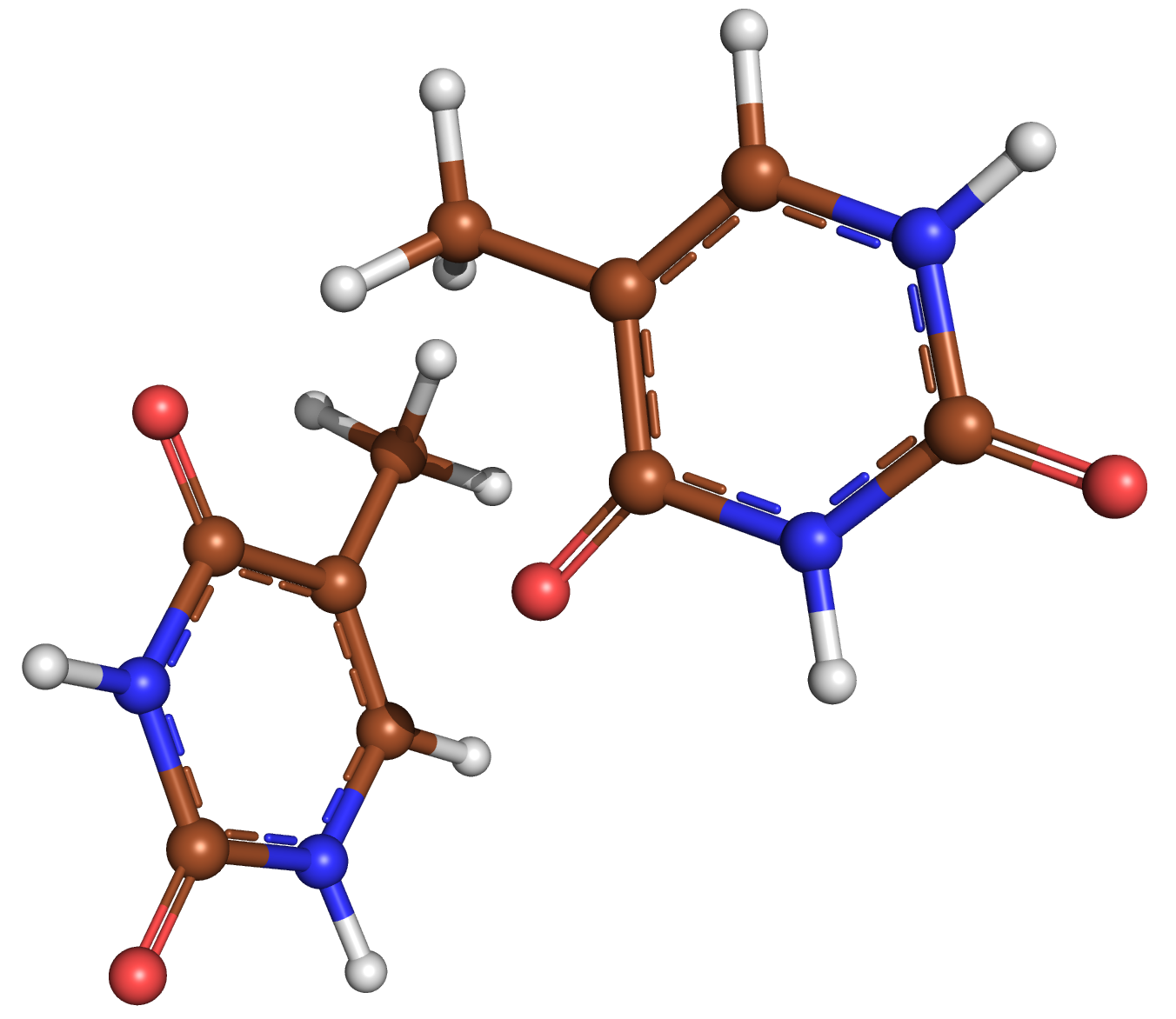
Рисунок 11. Оптимизированная структура при заряде системы 0. Произошел переход димера в тимины.
td1.out -3273.58217 EV
td2.out -3253.90834 EV
td3.out -3273.69661 EV.
Таким образом, энергии исходного и конечного состояния практически равны, хотя энергия конечного состояния примерно на 0.1 эв меньше (но сами значения энергий на 4 порядка больше). При добавлении электронов к возбужденному состоянию происходит переход димера в тимины, при этом происходит образование двух ароматических структур. Такая ситуация должна быть энергетически более выгодной, чем формирование димера (еще и с напряженной пространственной структурой). Однако разница в энергии этих состояний невелика, и этим можно объяснить возможность образования тиминовых димеров при облучении УФ.
© Наталья Ланина
e-mail: n.lanina@fbb.msu.ru
последний раз обновлялось: 12.3.15